

Screening for functional regulatory variants in open chromatin using GenIE-ATAC
- PMID: 37125635
- PMCID: PMC10287956
- DOI: 10.1093/nar/gkad332
Screening for functional regulatory variants in open chromatin using GenIE-ATAC
Abstract
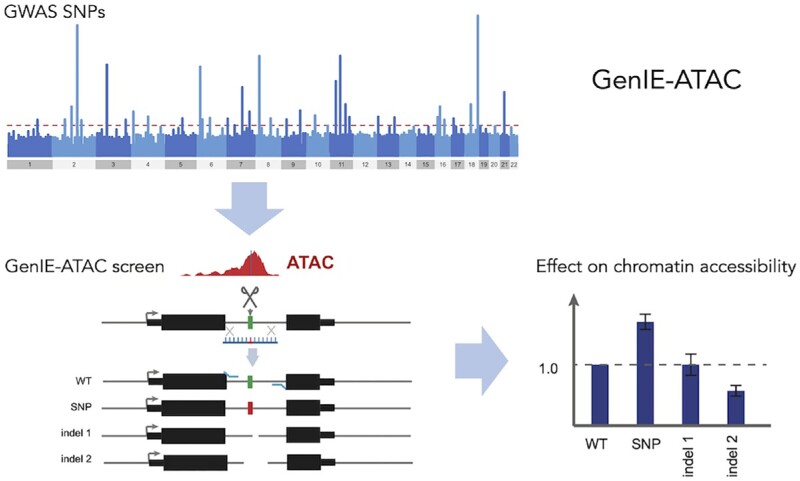
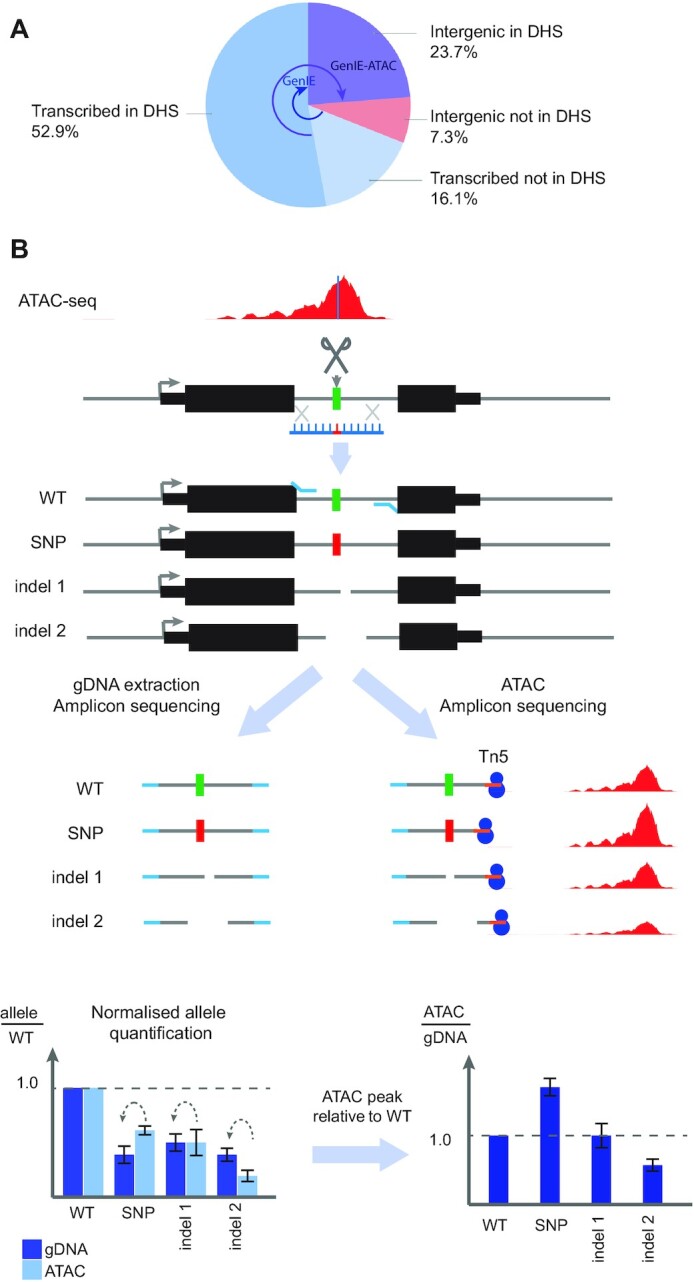
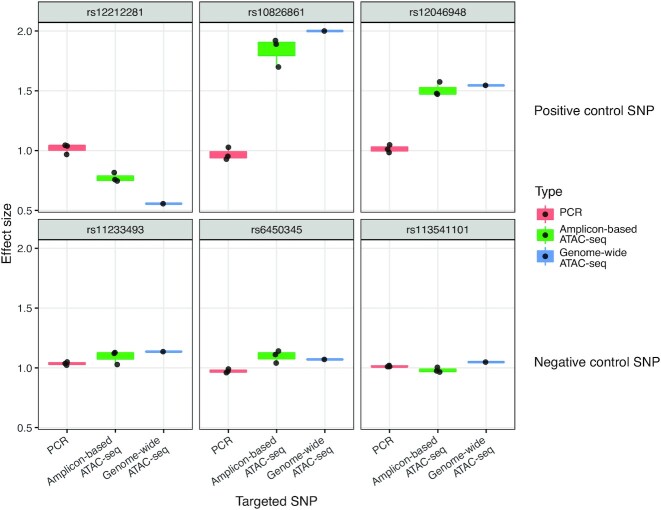
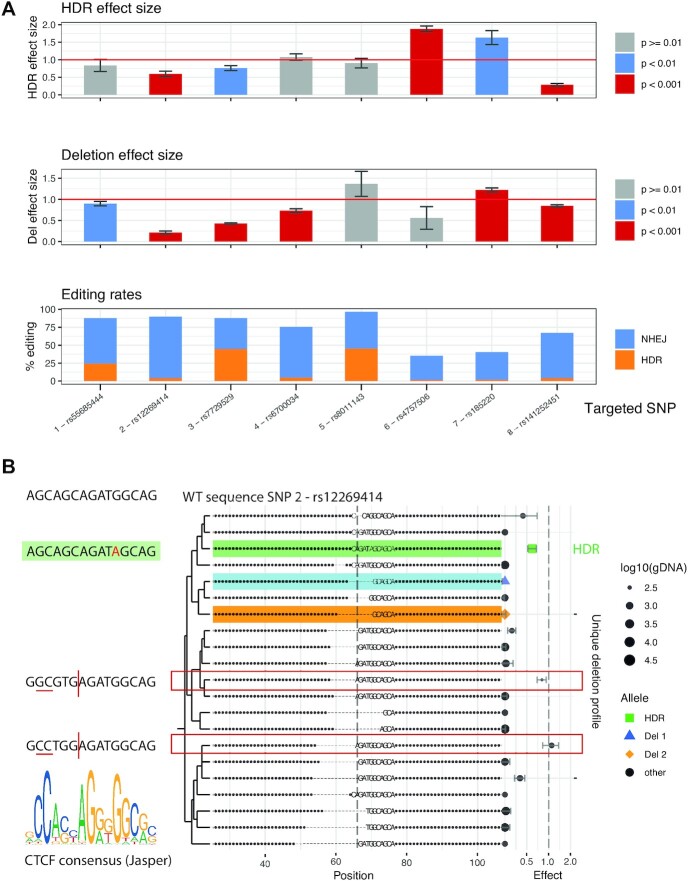
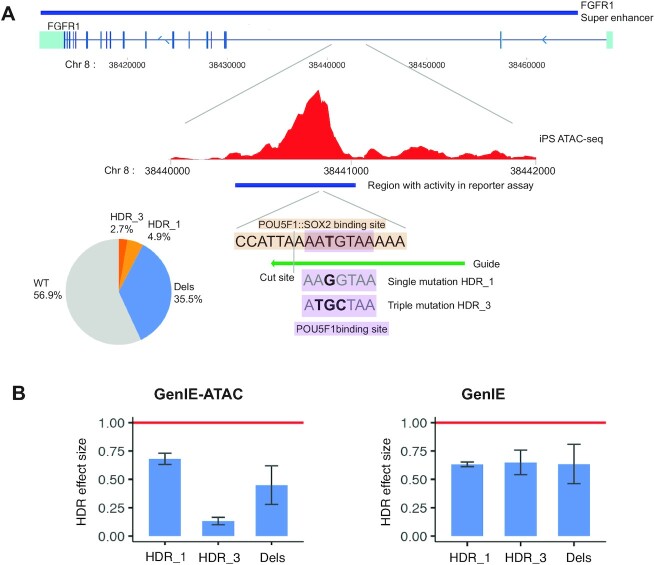
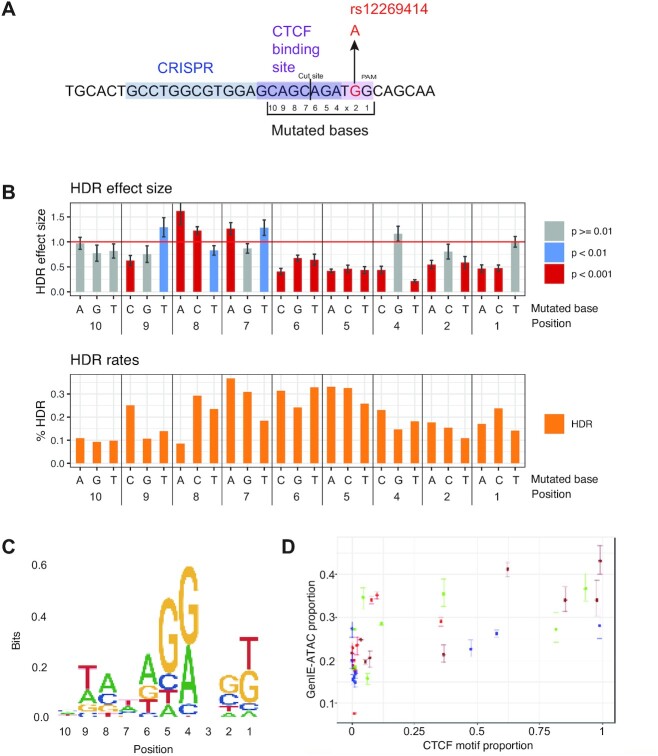
Understanding the effects of genetic variation in gene regulatory elements is crucial to interpreting genome function. This is particularly pertinent for the hundreds of thousands of disease-associated variants identified by GWAS, which frequently sit within gene regulatory elements but whose functional effects are often unknown. Current methods are limited in their scalability and ability to assay regulatory variants in their endogenous context, independently of other tightly linked variants. Here, we present a new medium-throughput screening system: genome engineering based interrogation of enhancers assay for transposase accessible chromatin (GenIE-ATAC), that measures the effect of individual variants on chromatin accessibility in their endogenous genomic and chromatin context. We employ this assay to screen for the effects of regulatory variants in human induced pluripotent stem cells, validating a subset of causal variants, and extend our software package (rgenie) to analyse these new data. We demonstrate that this methodology can be used to understand the impact of defined deletions and point mutations within transcription factor binding sites. We thus establish GenIE-ATAC as a method to screen for the effect of gene regulatory element variation, allowing identification and prioritisation of causal variants from GWAS for functional follow-up and understanding the mechanisms of regulatory element function.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E.et al.. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019; 47:D1005–D1012. - PMC - PubMed
-
- Ghoussaini M., Mountjoy E., Carmona M., Peat G., Schmidt E.M., Hercules A., Fumis L., Miranda A., Carvalho-Silva D., Buniello A.et al.. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 2021; 49:D1311–D1320. - PMC - PubMed
-
- Schwartzentruber J., Cooper S., Liu J.Z., Barrio-Hernandez I., Bello E., Kumasaka N., Johnson T., Estrada K., Gaffney D.J., Beltrao P.et al.. Genome-wide meta-analysis, fine-mapping, and integrative prioritization identify new Alzheimer's disease risk genes. Nat. Genet. 2021; 53:392–402. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
