

Biobank-scale inference of ancestral recombination graphs enables genealogical analysis of complex traits
- PMID: 37127670
- PMCID: PMC10181934
- DOI: 10.1038/s41588-023-01379-x
Biobank-scale inference of ancestral recombination graphs enables genealogical analysis of complex traits
Abstract
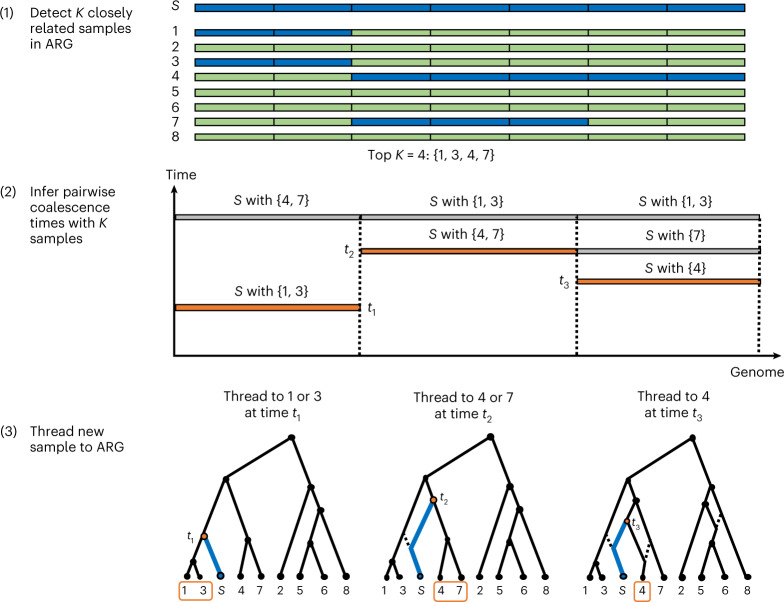
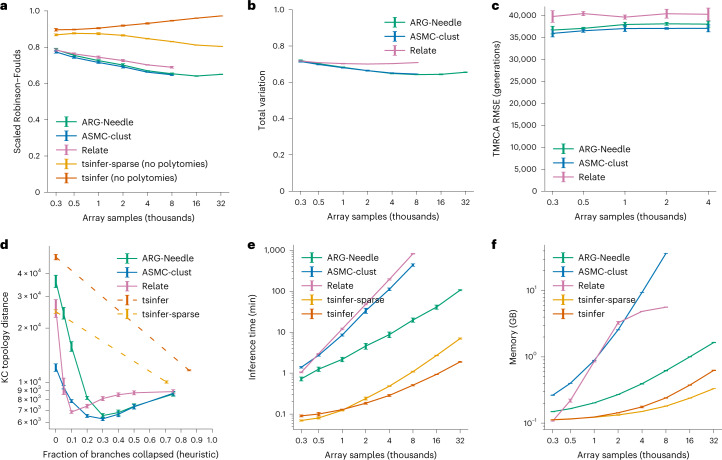
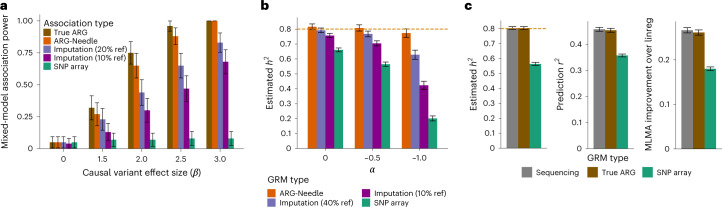
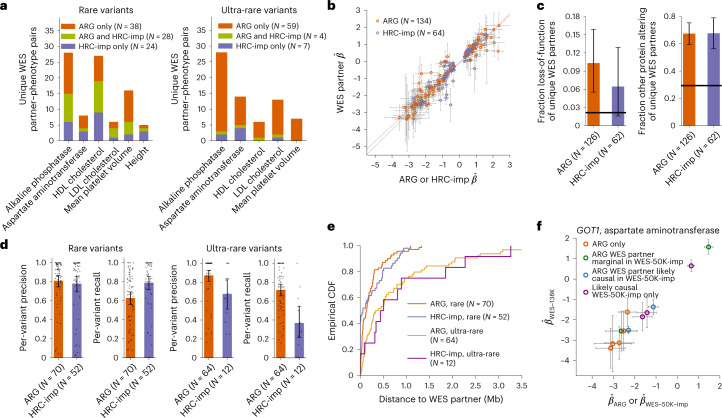
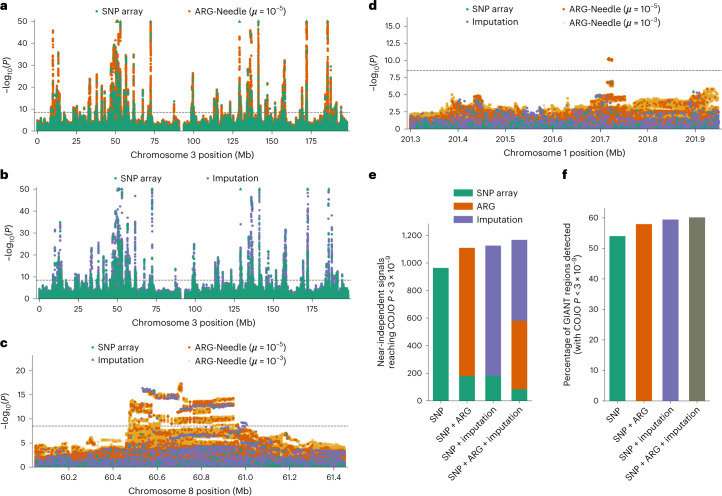
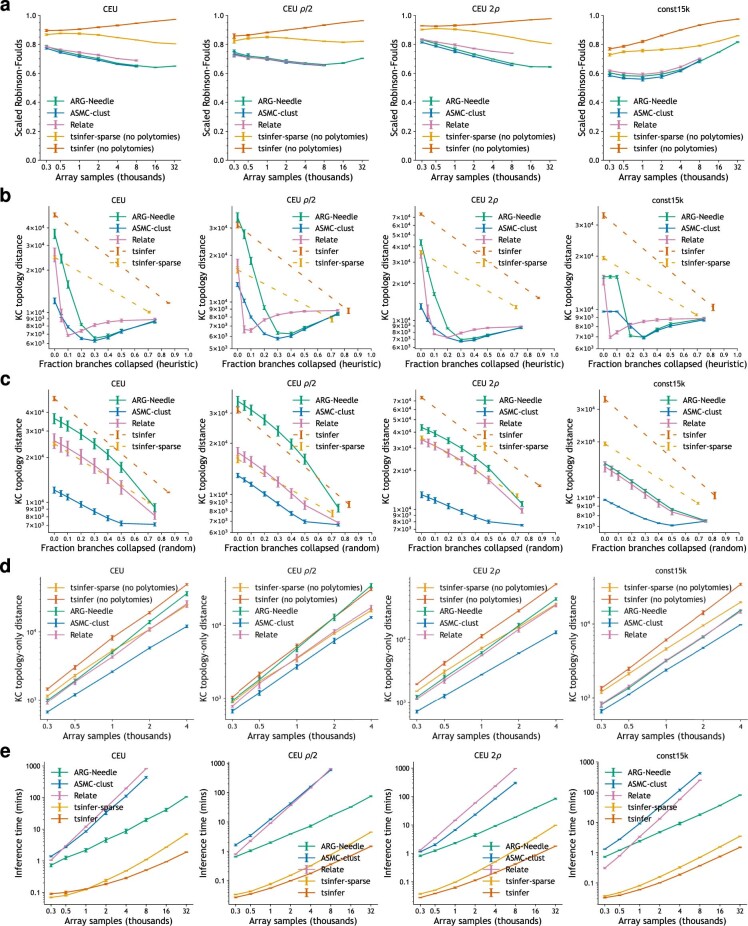
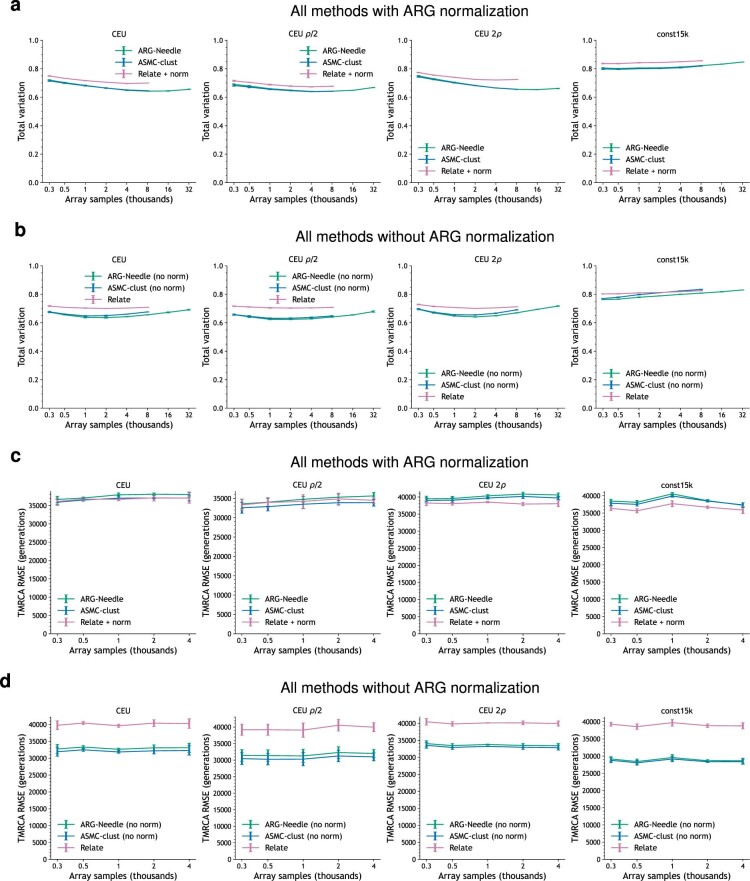
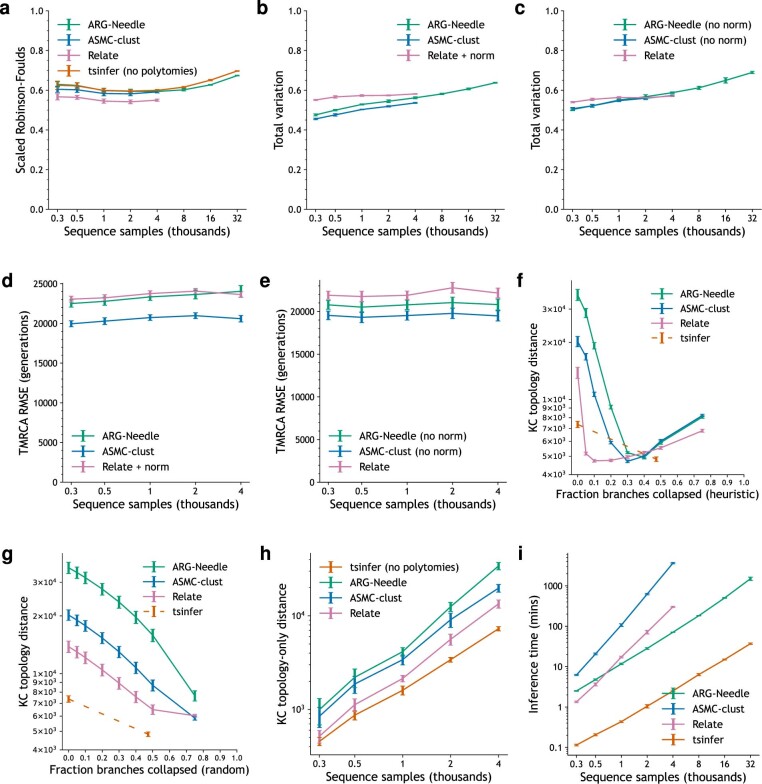
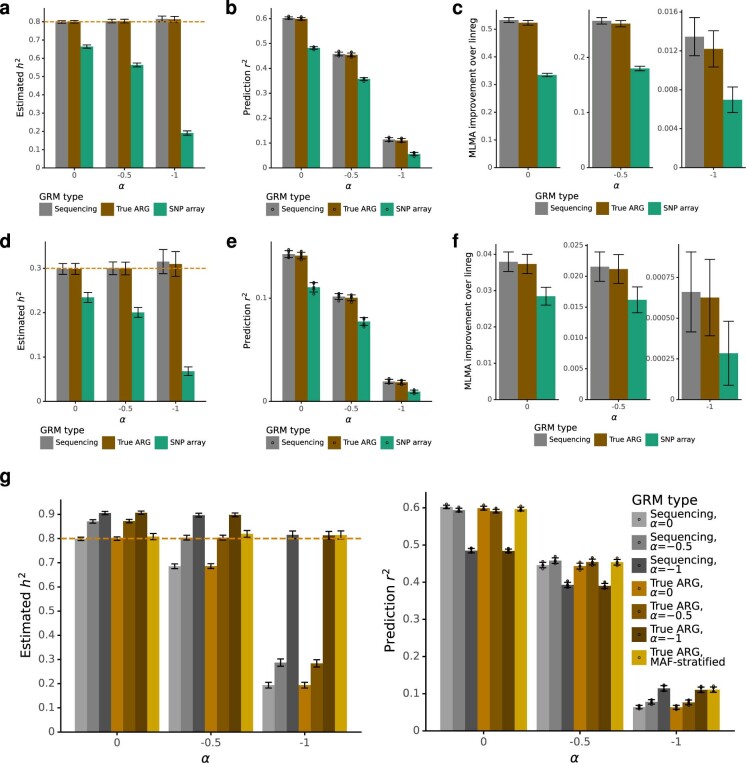
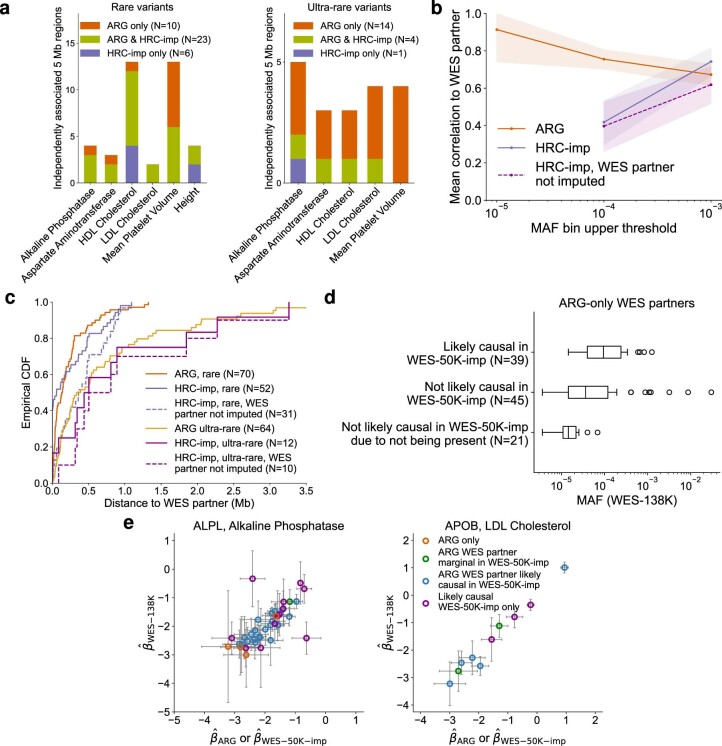
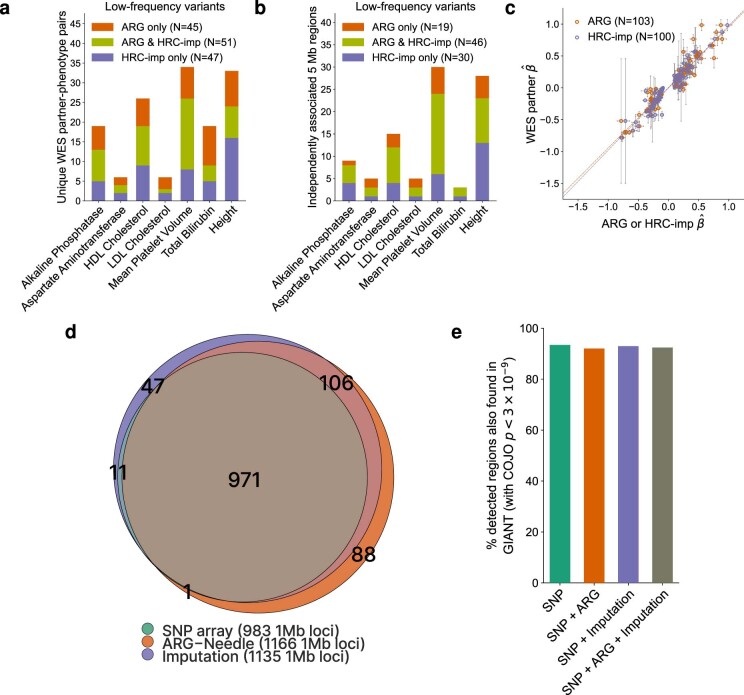
Genome-wide genealogies compactly represent the evolutionary history of a set of genomes and inferring them from genetic data has the potential to facilitate a wide range of analyses. We introduce a method, ARG-Needle, for accurately inferring biobank-scale genealogies from sequencing or genotyping array data, as well as strategies to utilize genealogies to perform association and other complex trait analyses. We use these methods to build genome-wide genealogies using genotyping data for 337,464 UK Biobank individuals and test for association across seven complex traits. Genealogy-based association detects more rare and ultra-rare signals (N = 134, frequency range 0.0007-0.1%) than genotype imputation using ~65,000 sequenced haplotypes (N = 64). In a subset of 138,039 exome sequencing samples, these associations strongly tag (average r = 0.72) underlying sequencing variants enriched (4.8×) for loss-of-function variation. These results demonstrate that inferred genome-wide genealogies may be leveraged in the analysis of complex traits, complementing approaches that require the availability of large, population-specific sequencing panels.
© 2023. The Author(s).
Conflict of interest statement
During the revision of this manuscript, A.B. became an employee of 54gene and Á.F.G. became an employee of deCODE genetics/Amgen. The remaining authors declare no competing interests.
Figures
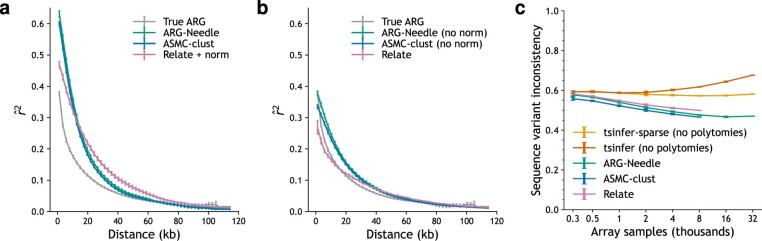
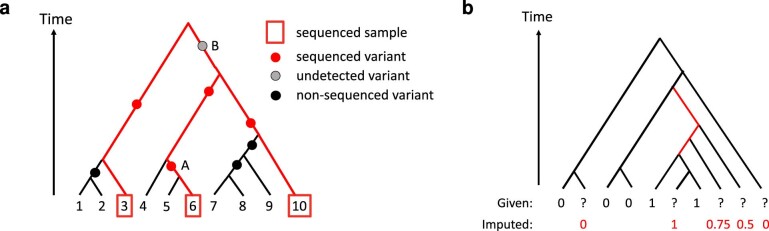
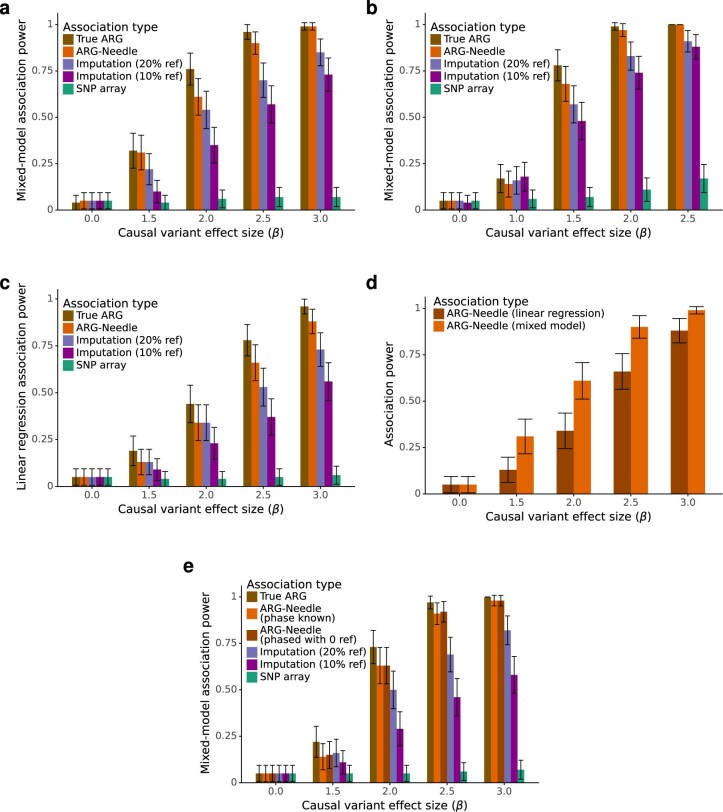
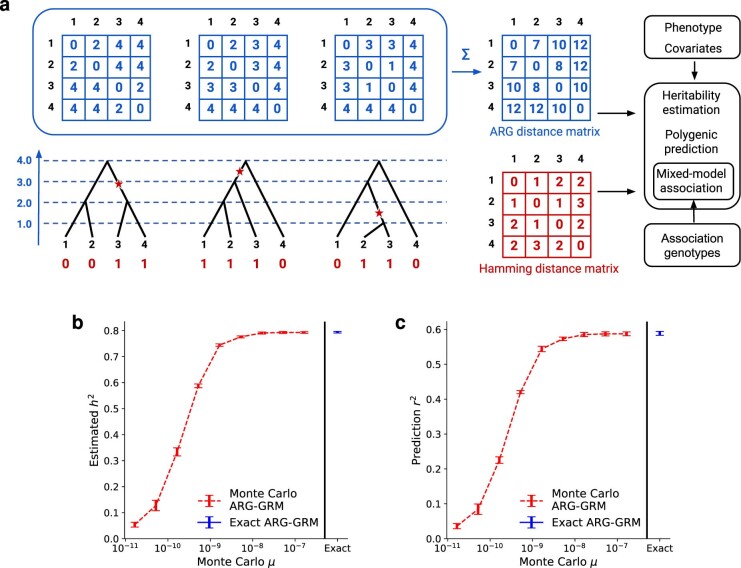
References
-
- Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat. Rev. Genet. 2003;4:99–110. - PubMed
-
- Beichman AC, Huerta-Sanchez E, Lohmueller KE. Using genomic data to infer historic population dynamics of nonmodel organisms. Annu. Rev. Ecol. Evol. Syst. 2018;49:433–456.
-
- Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
