

The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer
- PMID: 37127787
- PMCID: PMC10212769
- DOI: 10.1038/s43018-023-00548-5
The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer
Abstract
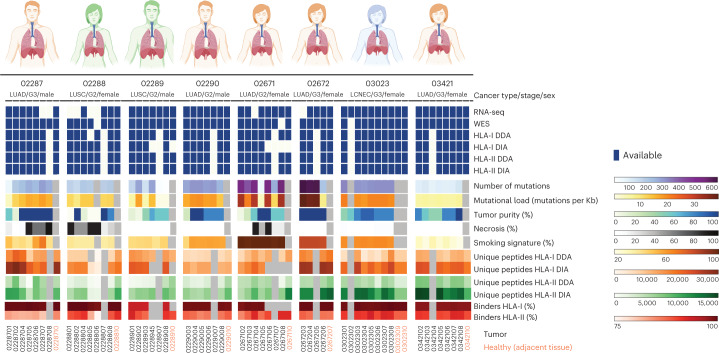
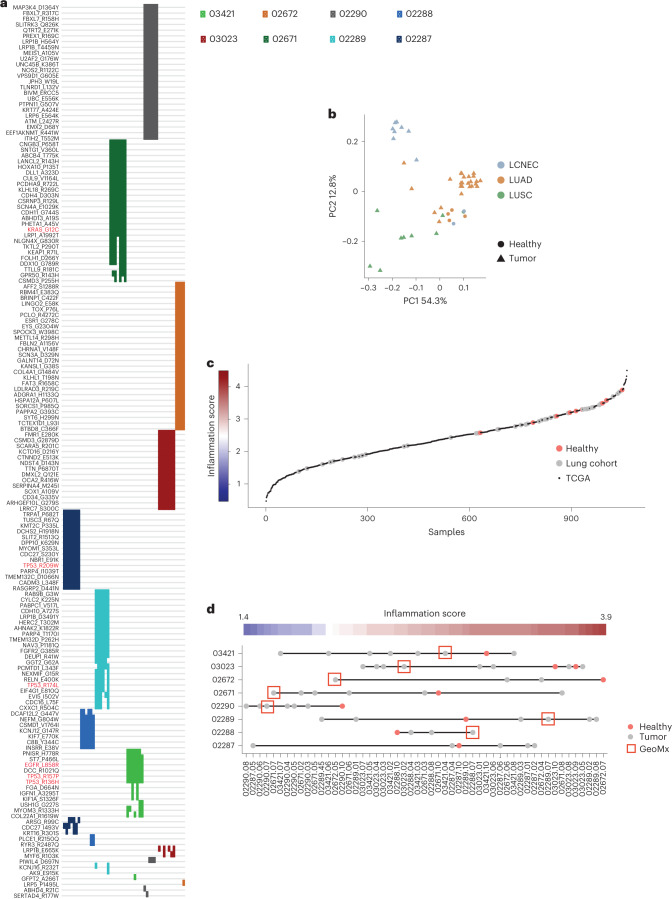
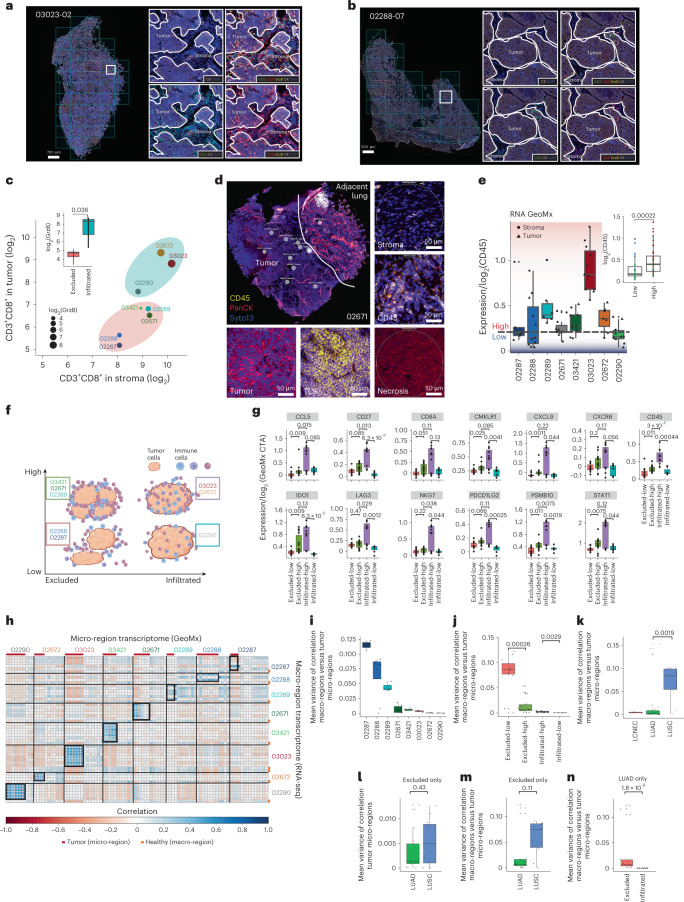
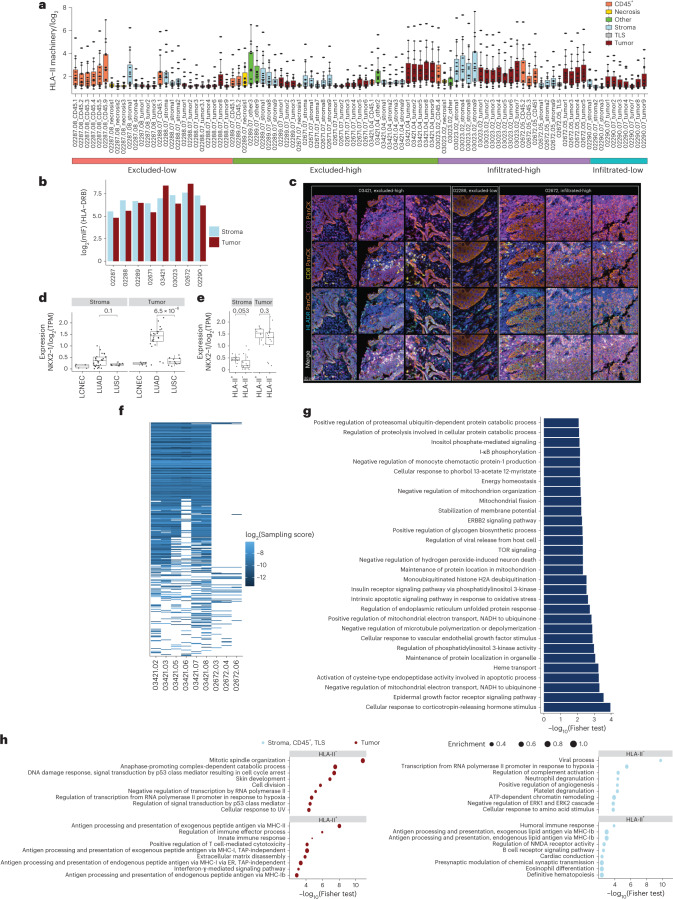
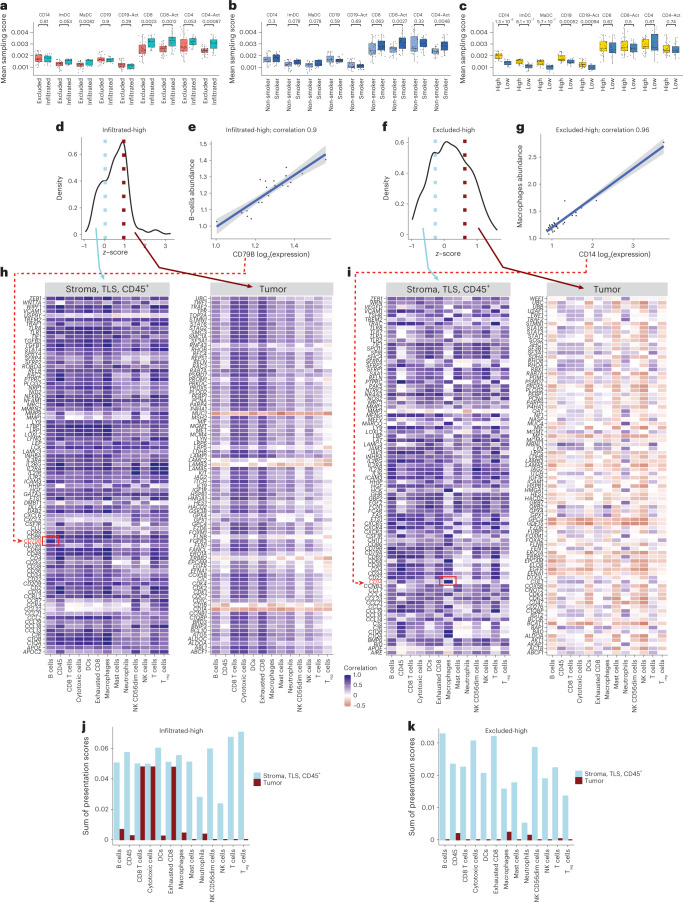
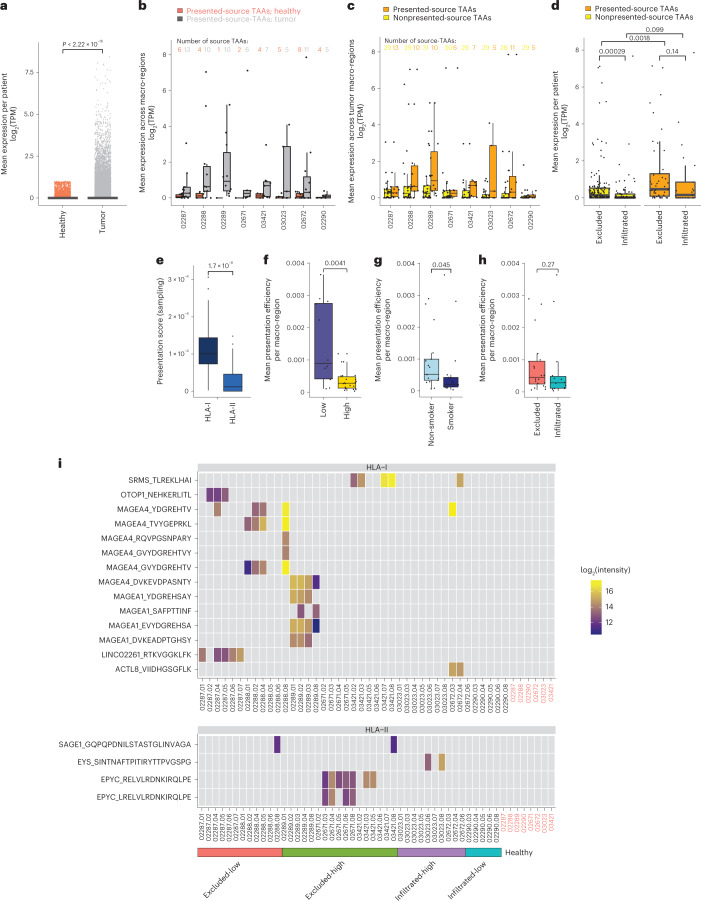
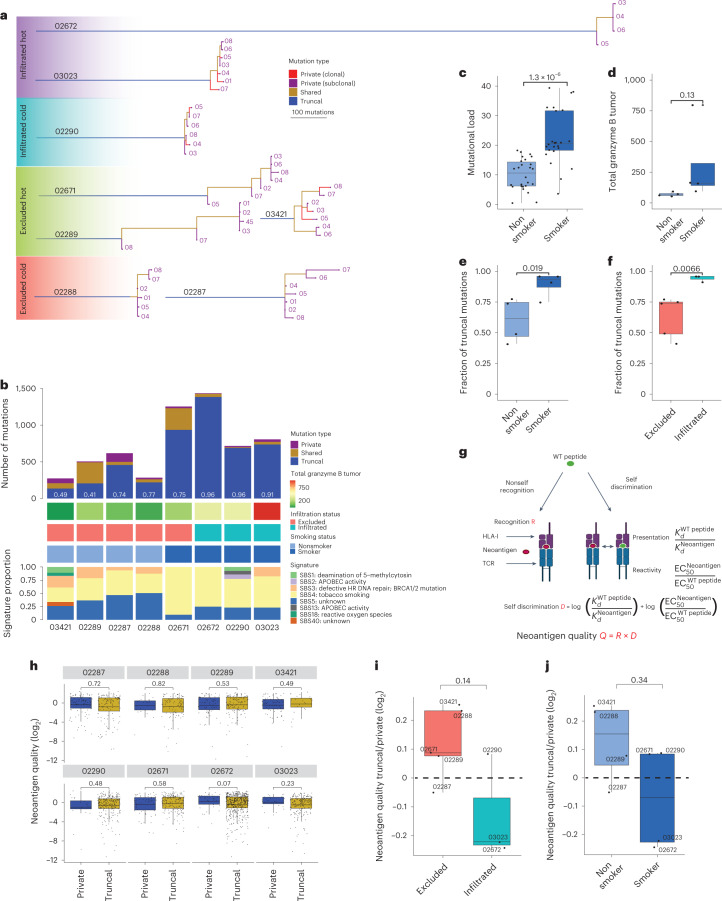
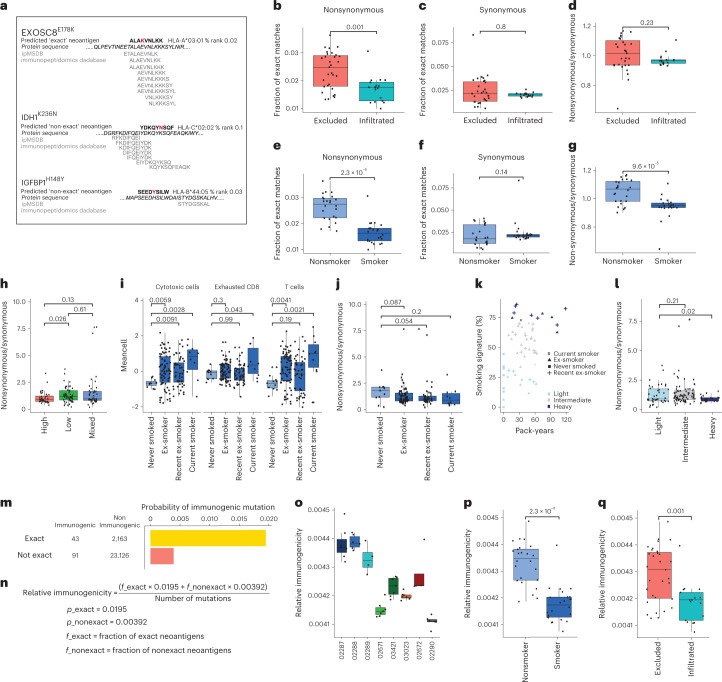
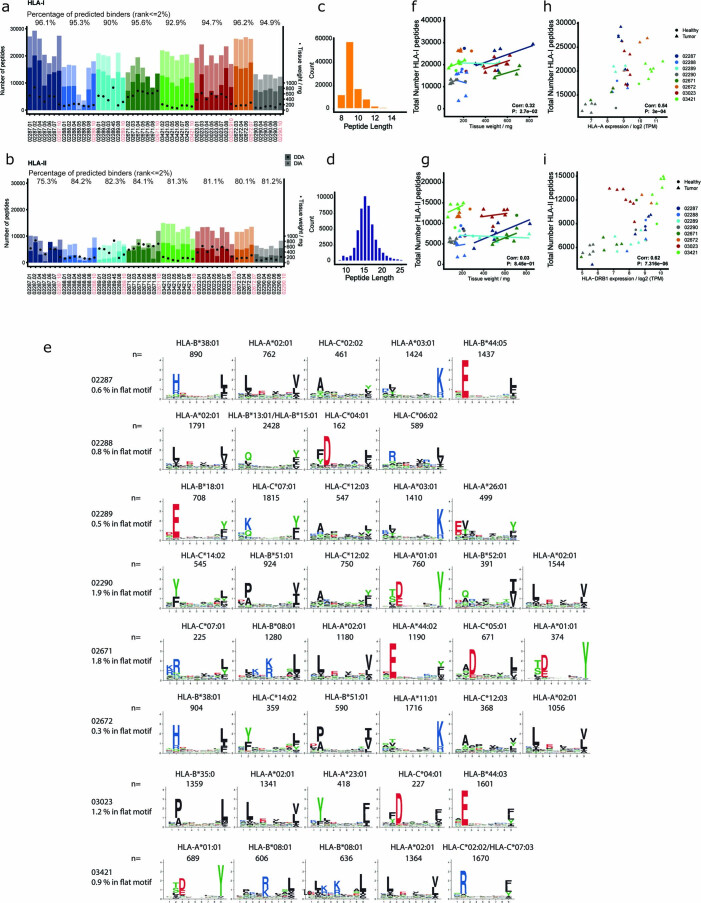
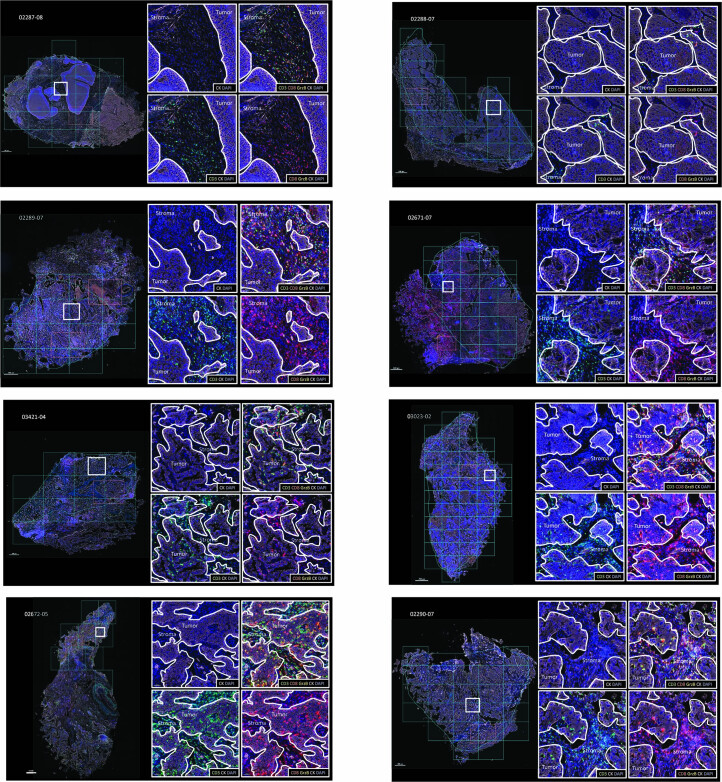
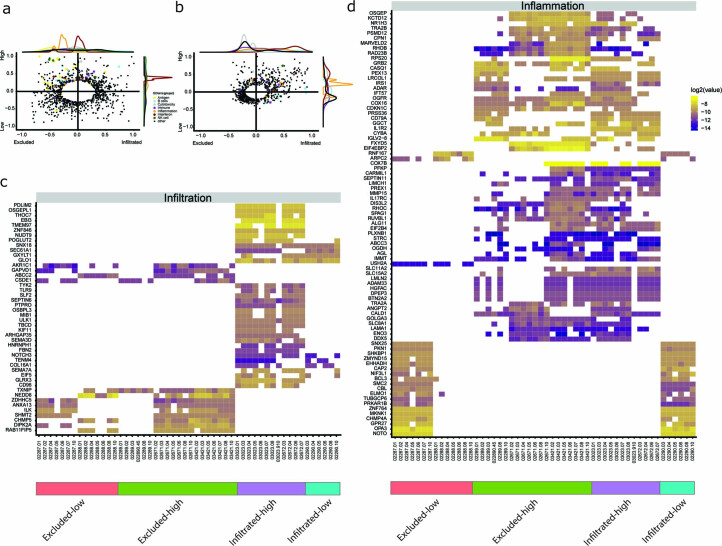
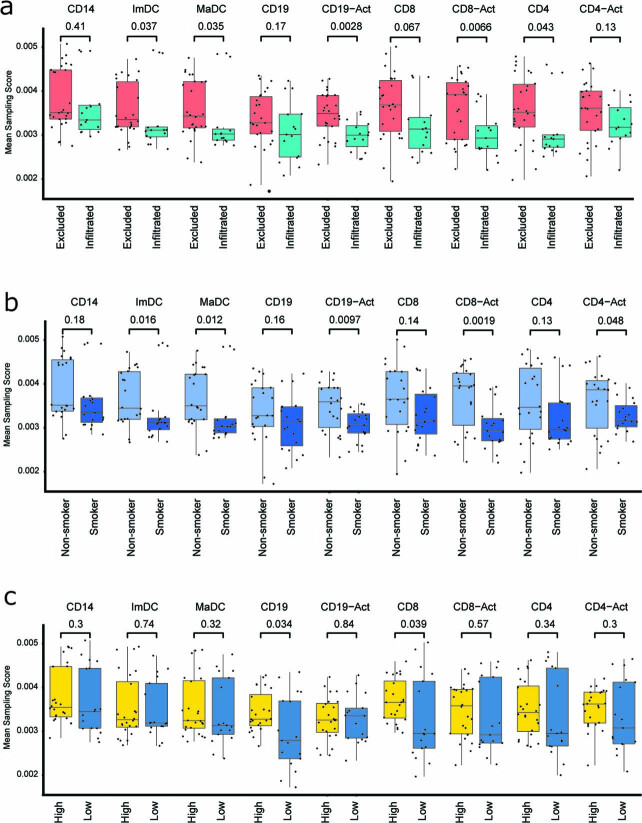
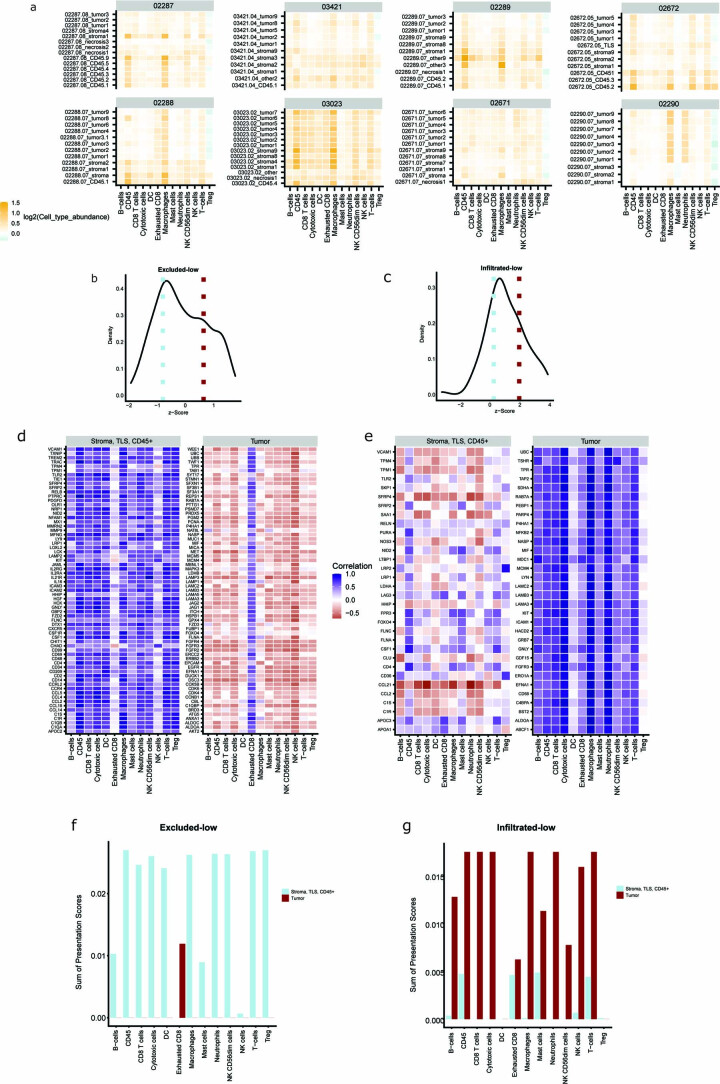
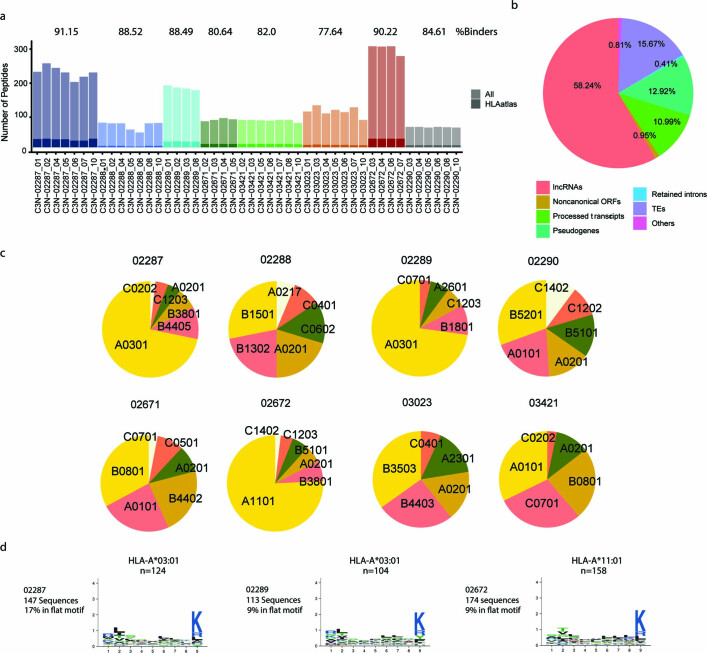
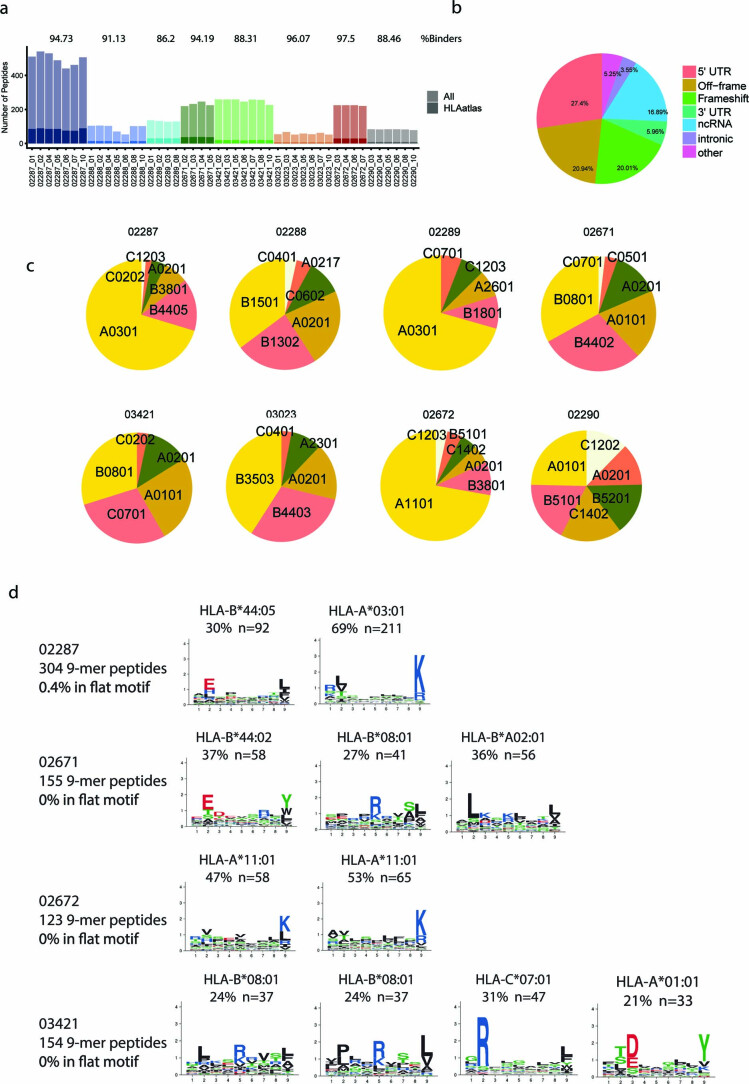
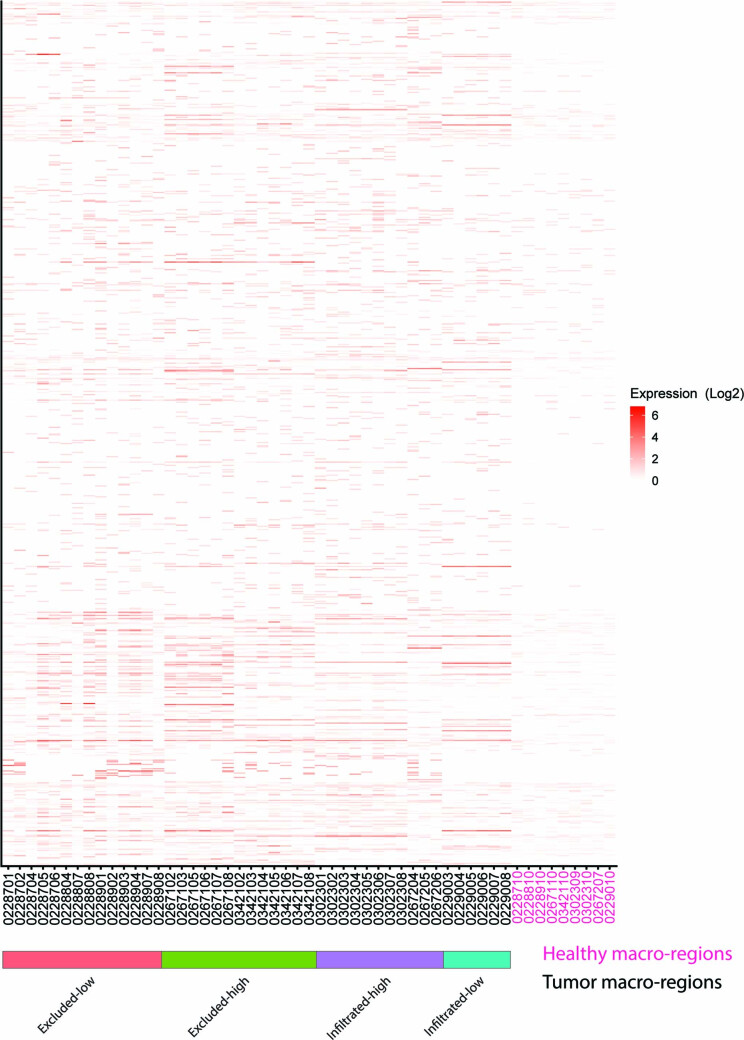
One key barrier to improving efficacy of personalized cancer immunotherapies that are dependent on the tumor antigenic landscape remains patient stratification. Although patients with CD3+CD8+ T cell-inflamed tumors typically show better response to immune checkpoint inhibitors, it is still unknown whether the immunopeptidome repertoire presented in highly inflamed and noninflamed tumors is substantially different. We surveyed 61 tumor regions and adjacent nonmalignant lung tissues from 8 patients with lung cancer and performed deep antigen discovery combining immunopeptidomics, genomics, bulk and spatial transcriptomics, and explored the heterogeneous expression and presentation of tumor (neo)antigens. In the present study, we associated diverse immune cell populations with the immunopeptidome and found a relatively higher frequency of predicted neoantigens located within HLA-I presentation hotspots in CD3+CD8+ T cell-excluded tumors. We associated such neoantigens with immune recognition, supporting their involvement in immune editing. This could have implications for the choice of combination therapies tailored to the patient's mutanome and immune microenvironment.
© 2023. The Author(s).
Conflict of interest statement
In the last 3 years, G.C. has received grants and research support or has been coinvestigator in clinical trials by Bristol-Myers Squibb, Tigen Pharma, Iovance, F. Hoffmann La Roche AG and Boehringer Ingelheim. The Lausanne University Hospital (CHUV) has received honoraria for advisory services that G.C. has provided to Genentech, AstraZeneca AG and EVIR. Patent WO2019086711A1 related to the NeoTIL technology from the Coukos laboratory has been licensed by the Ludwig Institute, also on behalf of the University of Lausanne and the CHUV, to Tigen Pharma. G.C. has previously received royalties from the University of Pennsylvania for CAR-T cell therapy licensed to Novartis and Tmunity Therapeutics. The remaining authors declare no competing interests.
Figures


Comment in
-
Taking the temperature of lung cancer antigens.Nat Cancer. 2023 May;4(5):586-587. doi: 10.1038/s43018-023-00552-9. Nat Cancer. 2023. PMID: 37237079 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
