

Conserved transcriptional connectivity of regulatory T cells in the tumor microenvironment informs new combination cancer therapy strategies
- PMID: 37127830
- PMCID: PMC10232368
- DOI: 10.1038/s41590-023-01504-2
Conserved transcriptional connectivity of regulatory T cells in the tumor microenvironment informs new combination cancer therapy strategies
Abstract
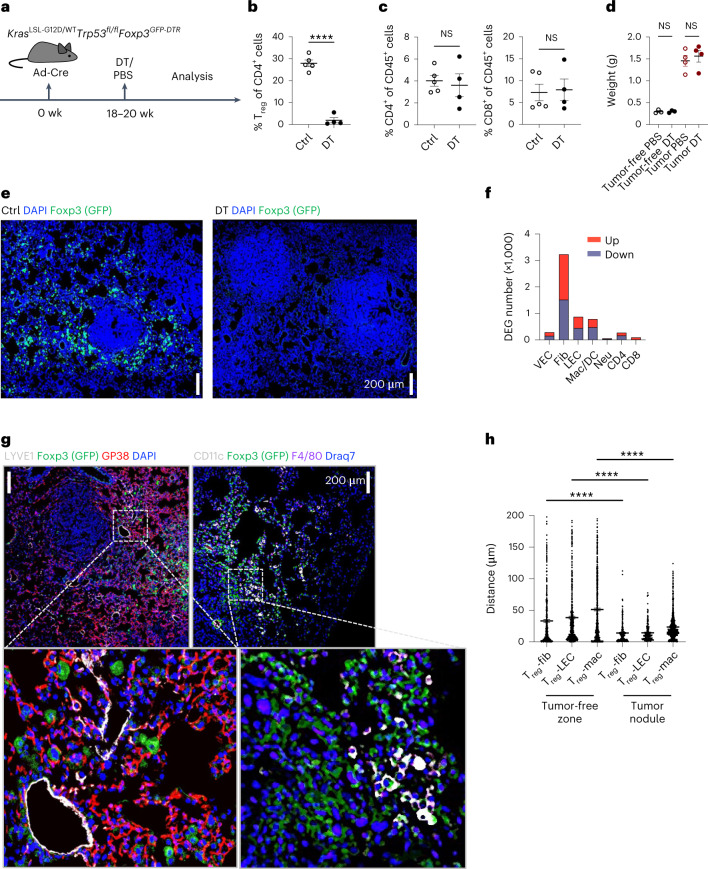
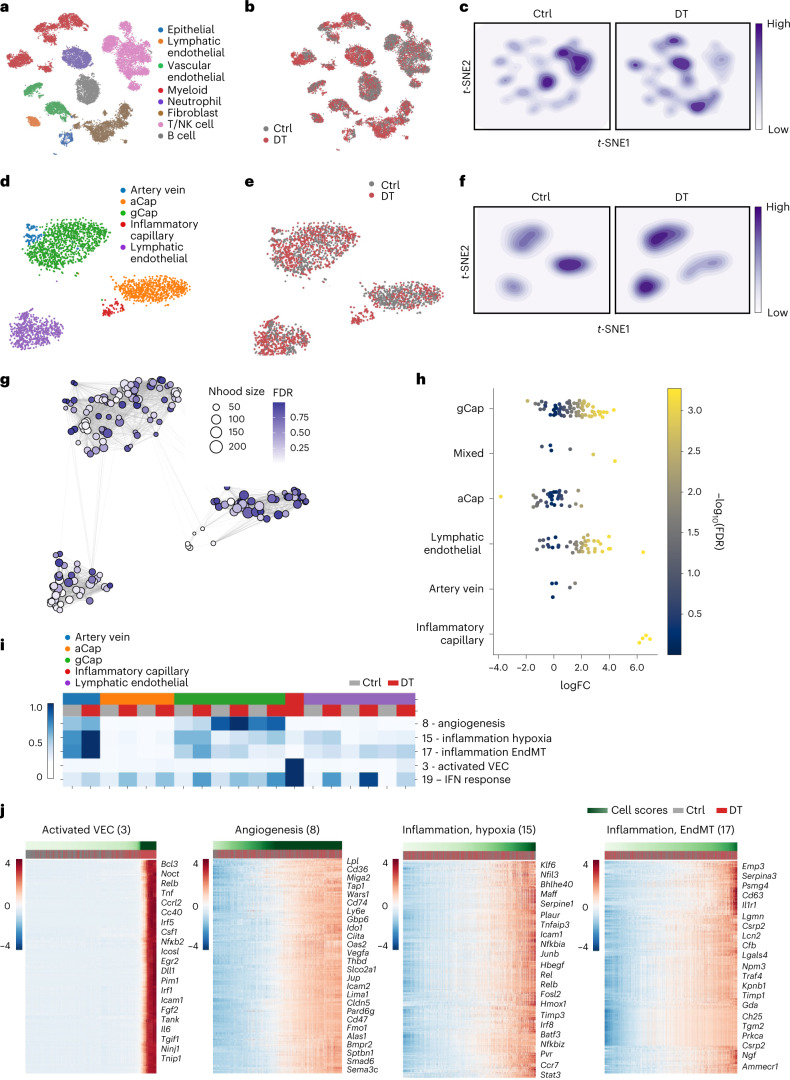
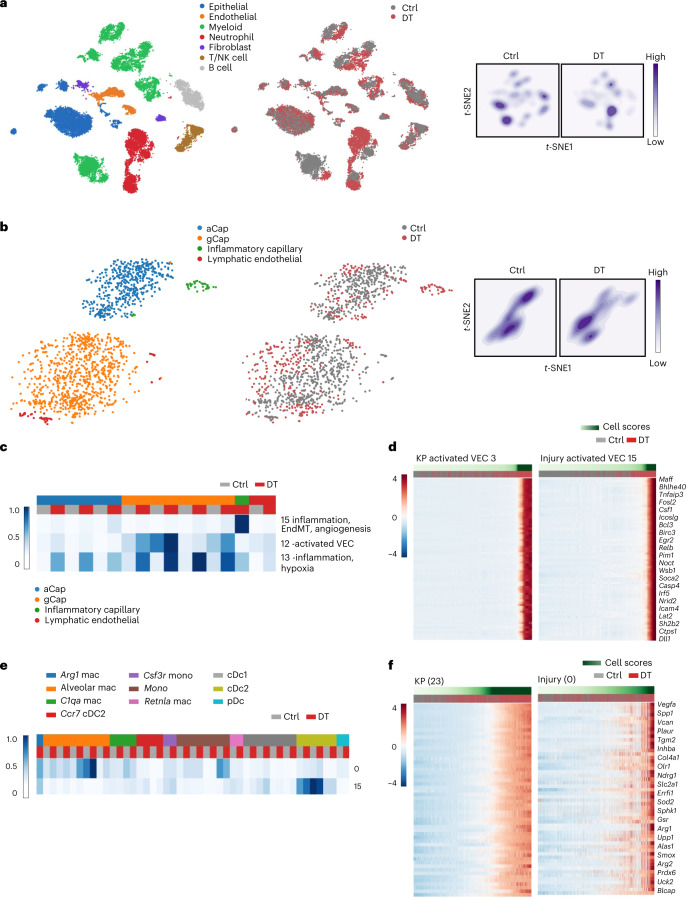
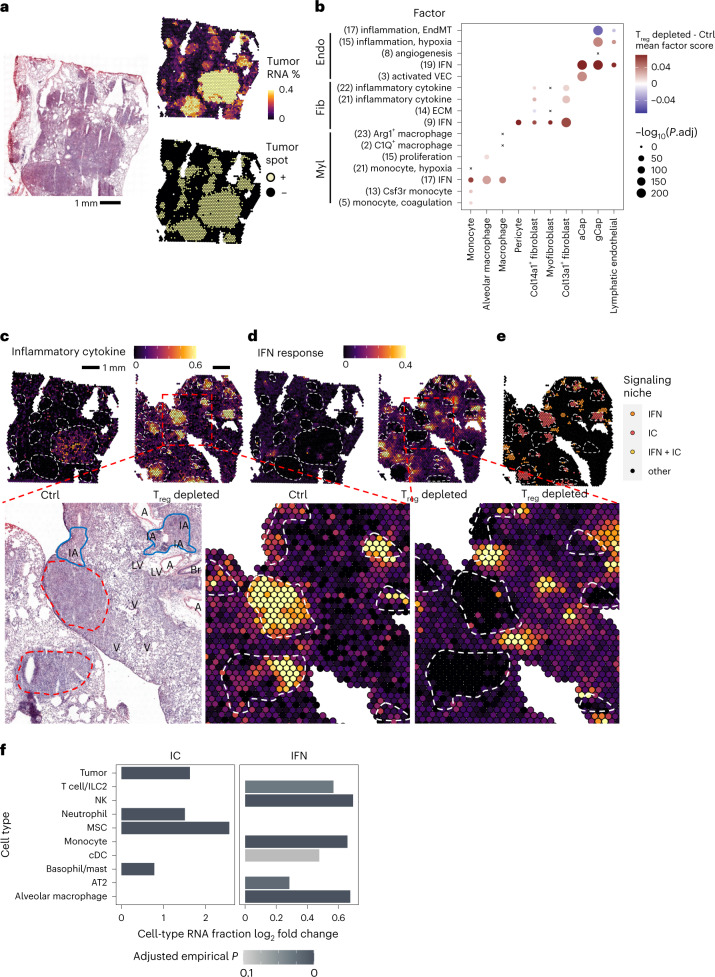
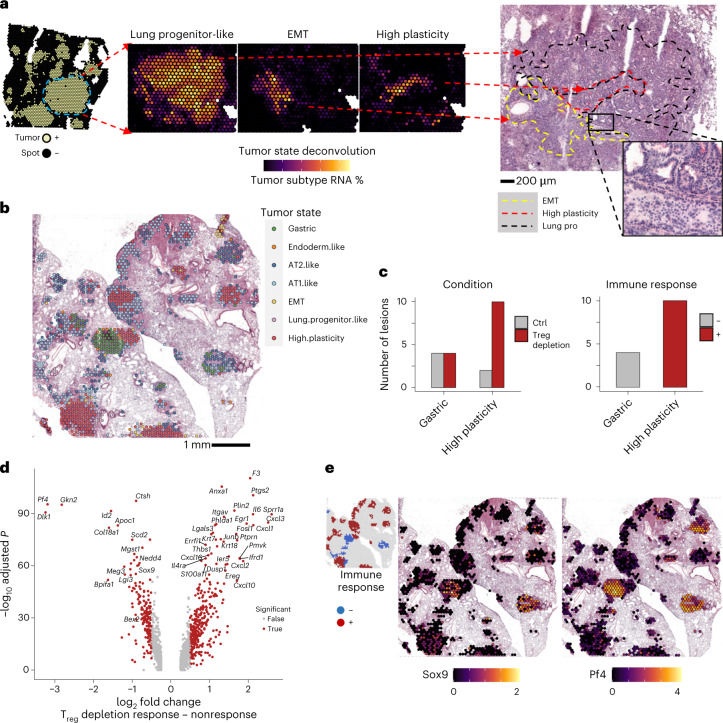
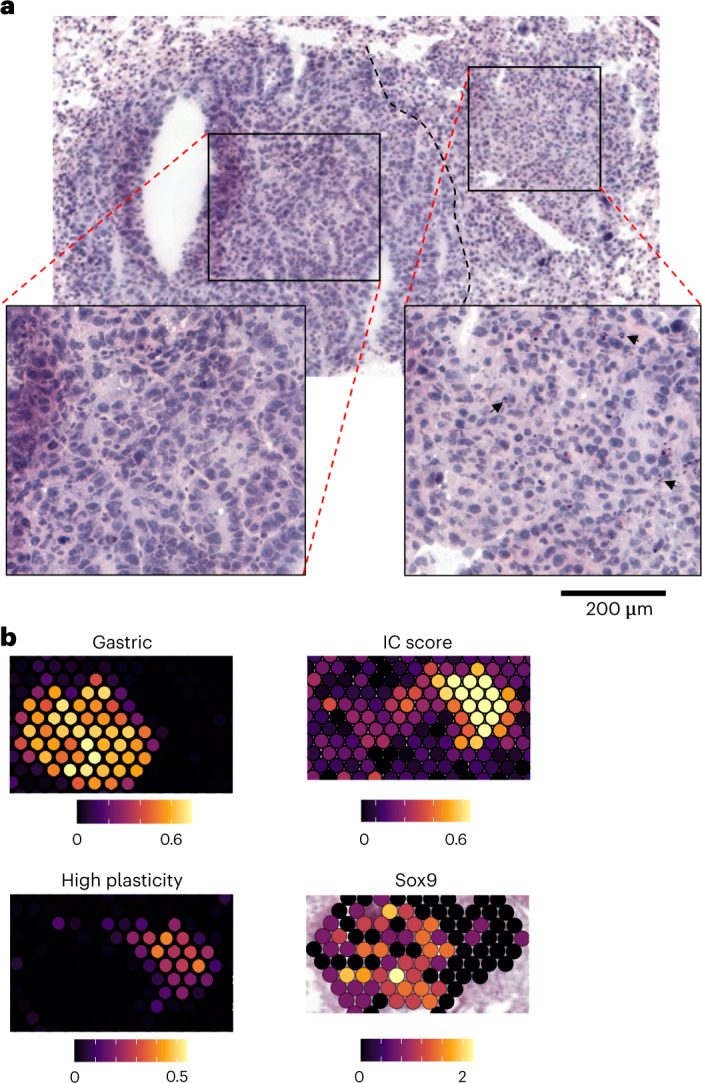
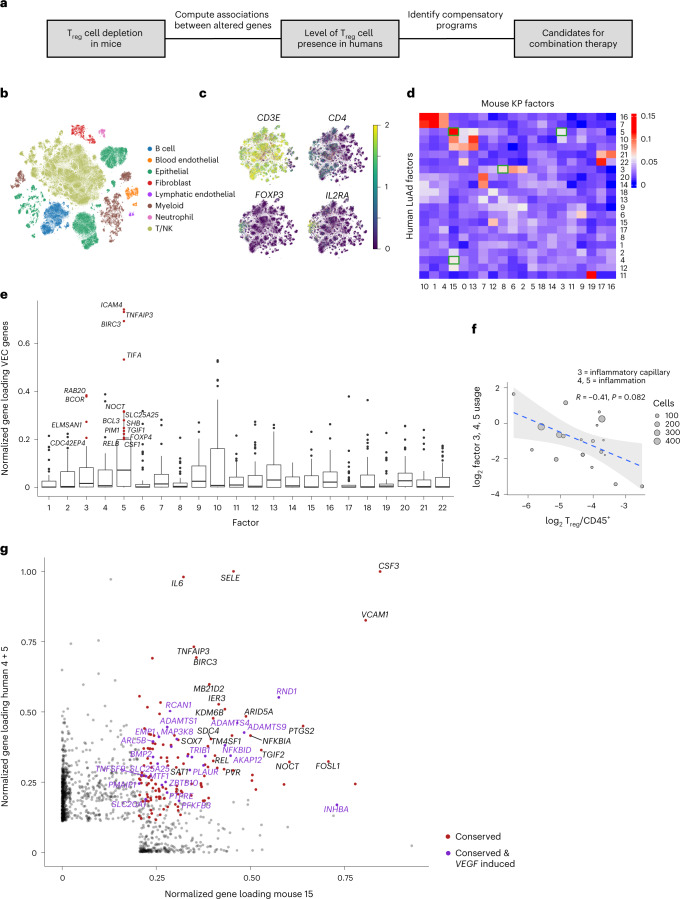
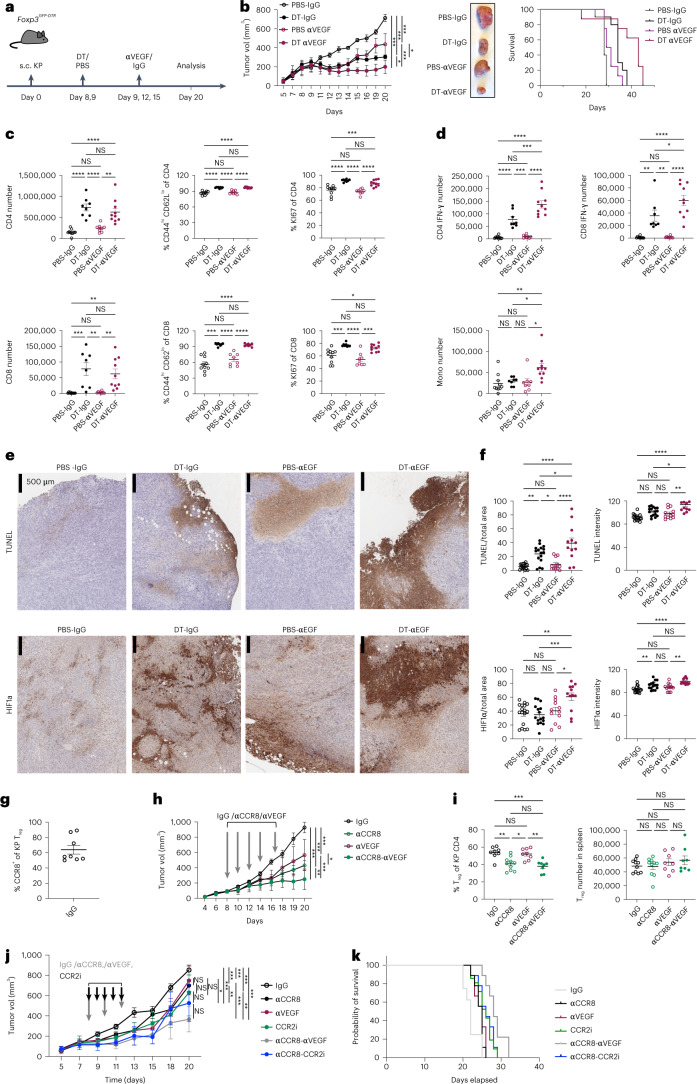
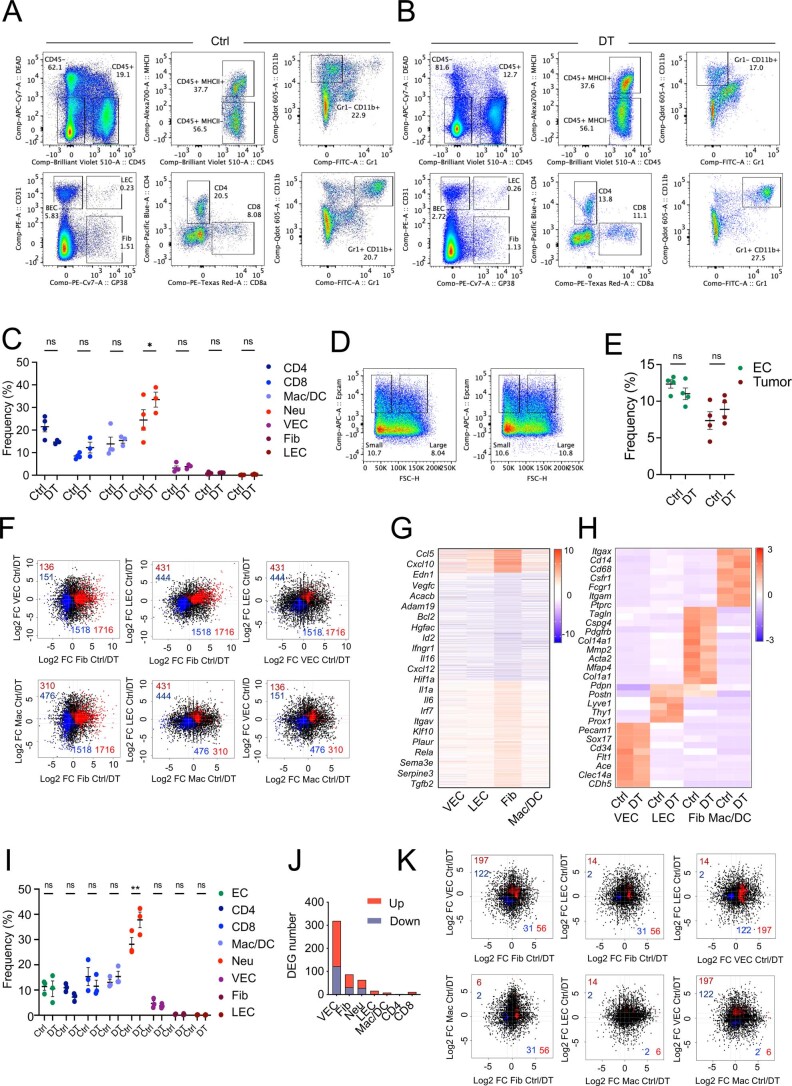
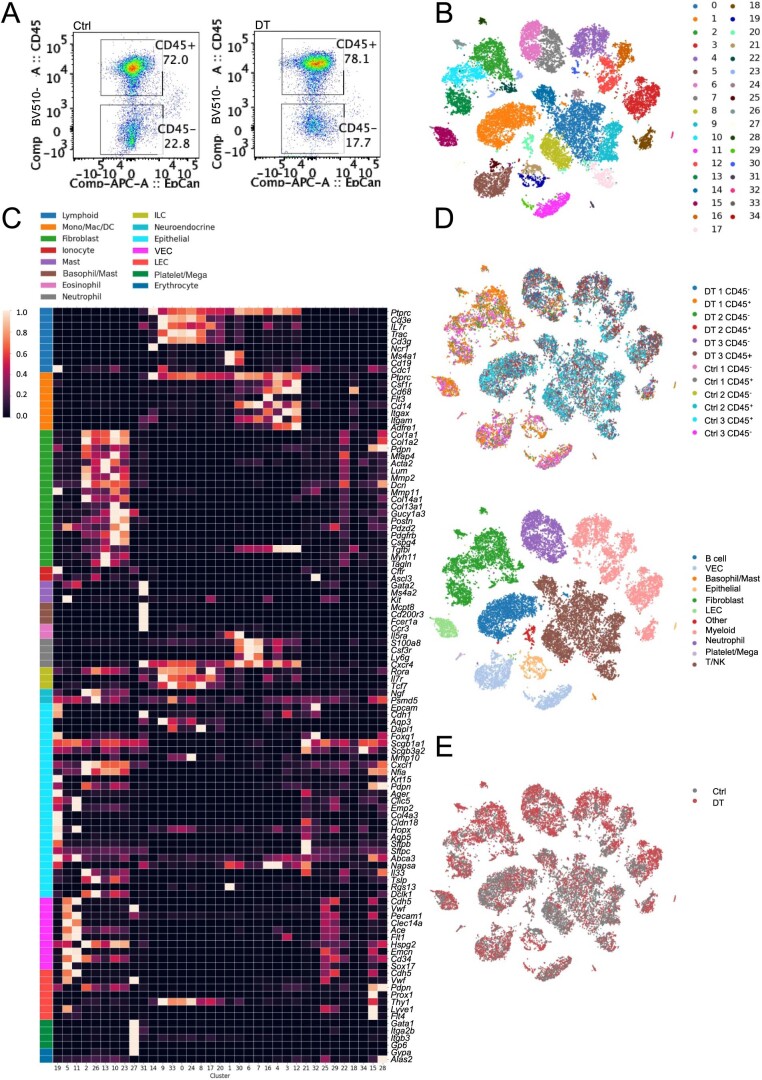
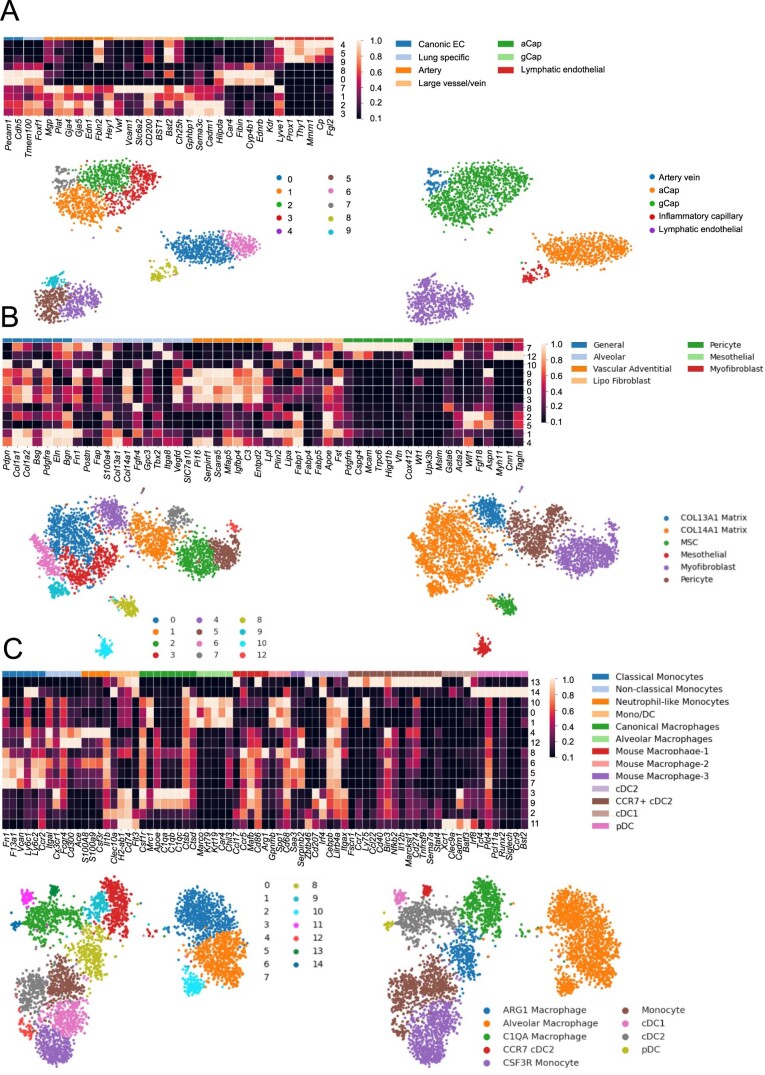
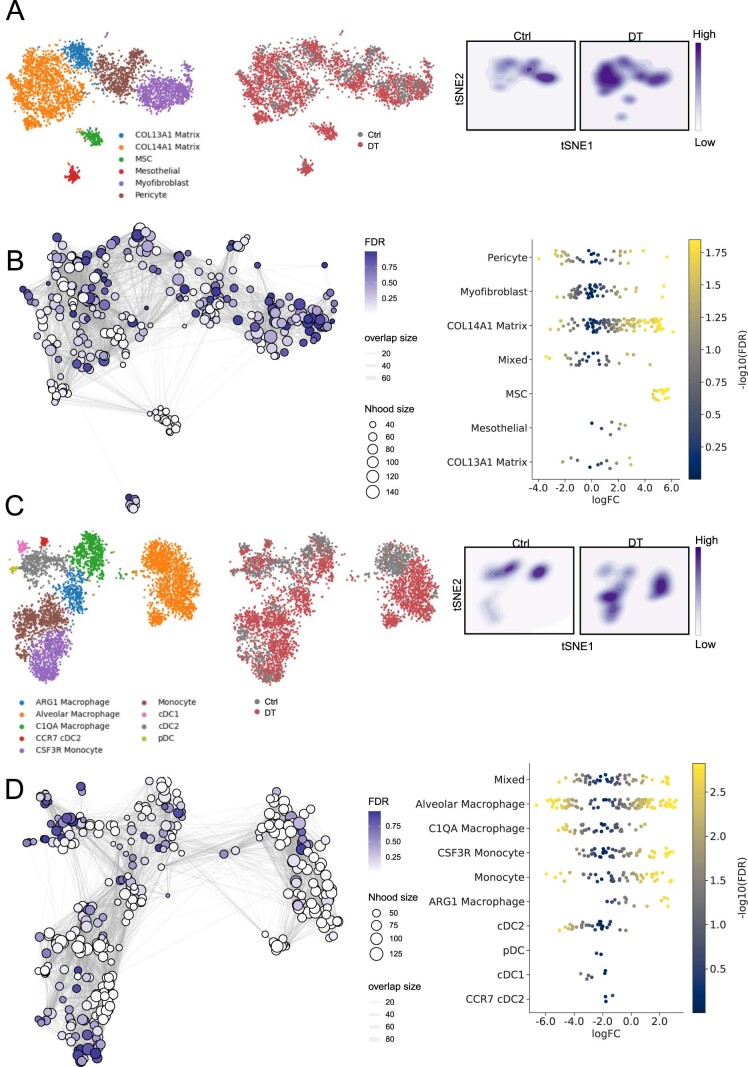
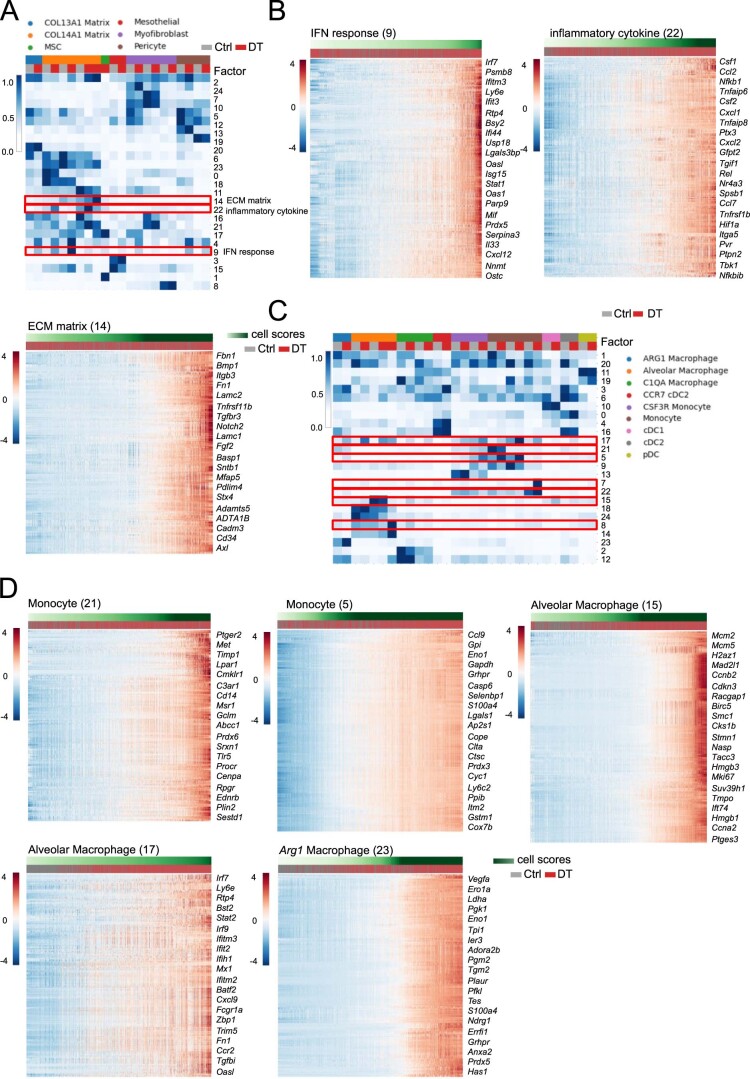
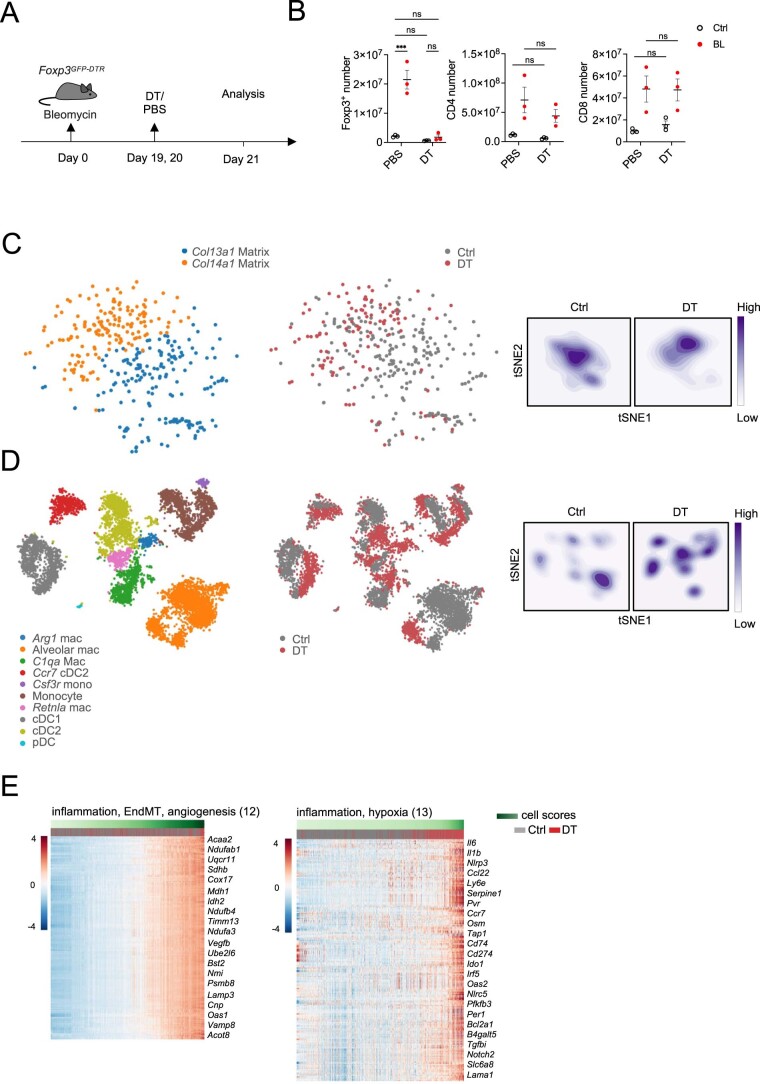
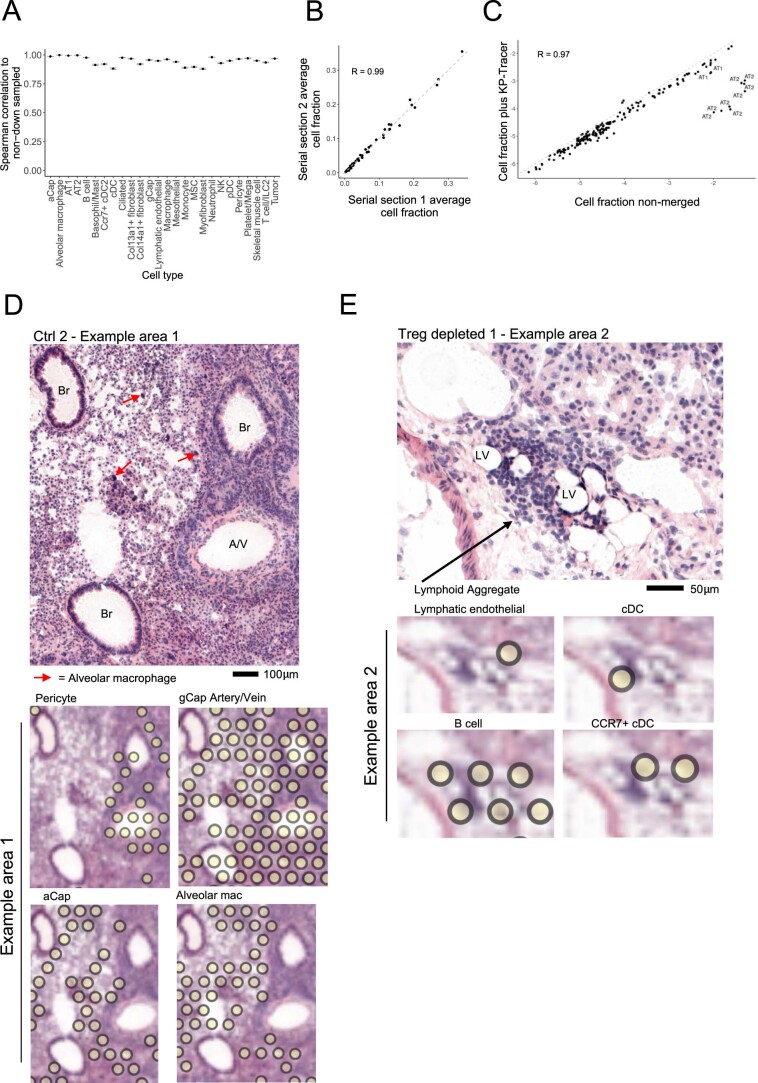
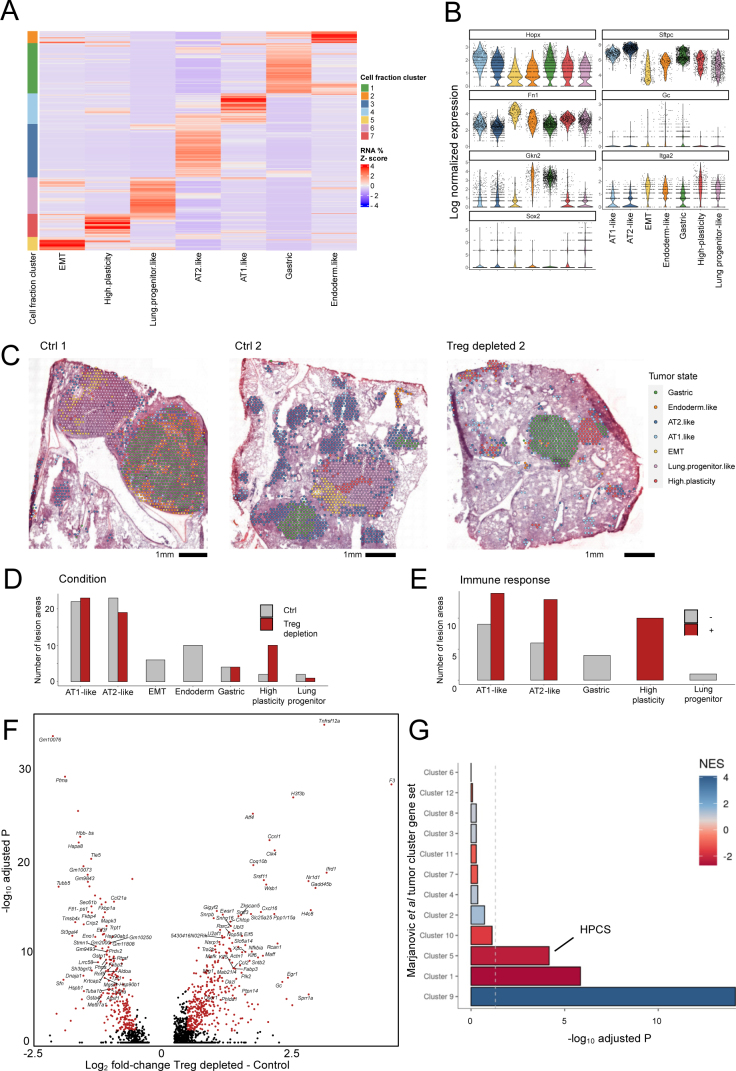
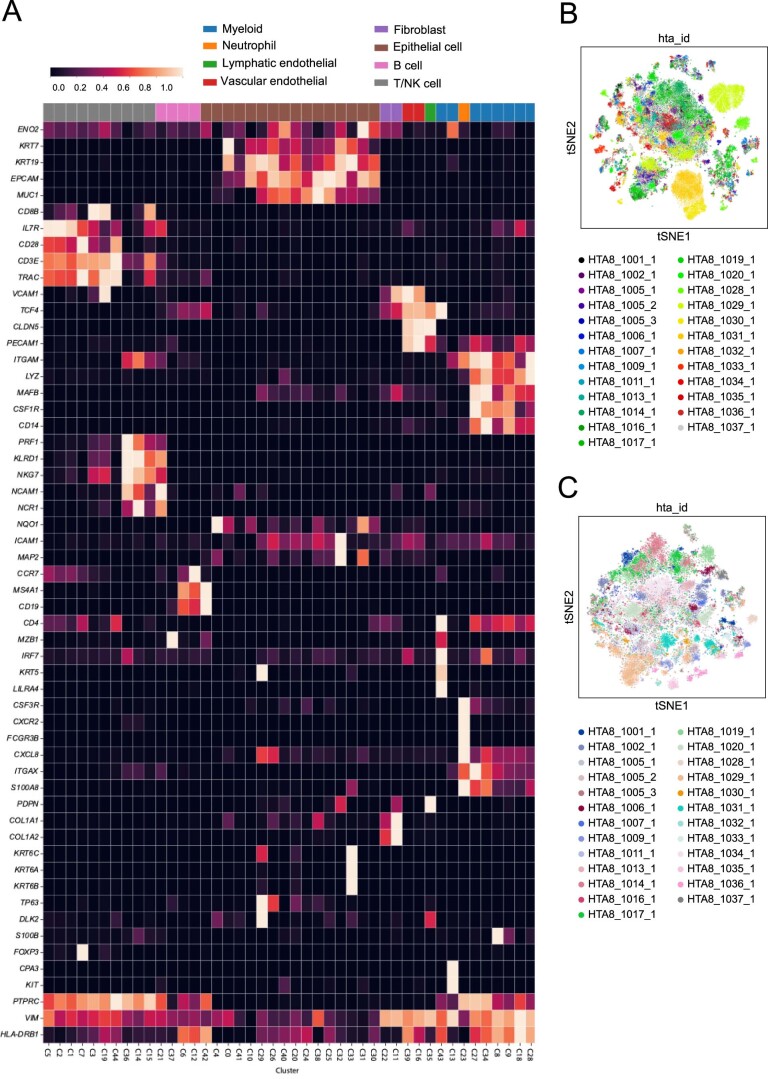
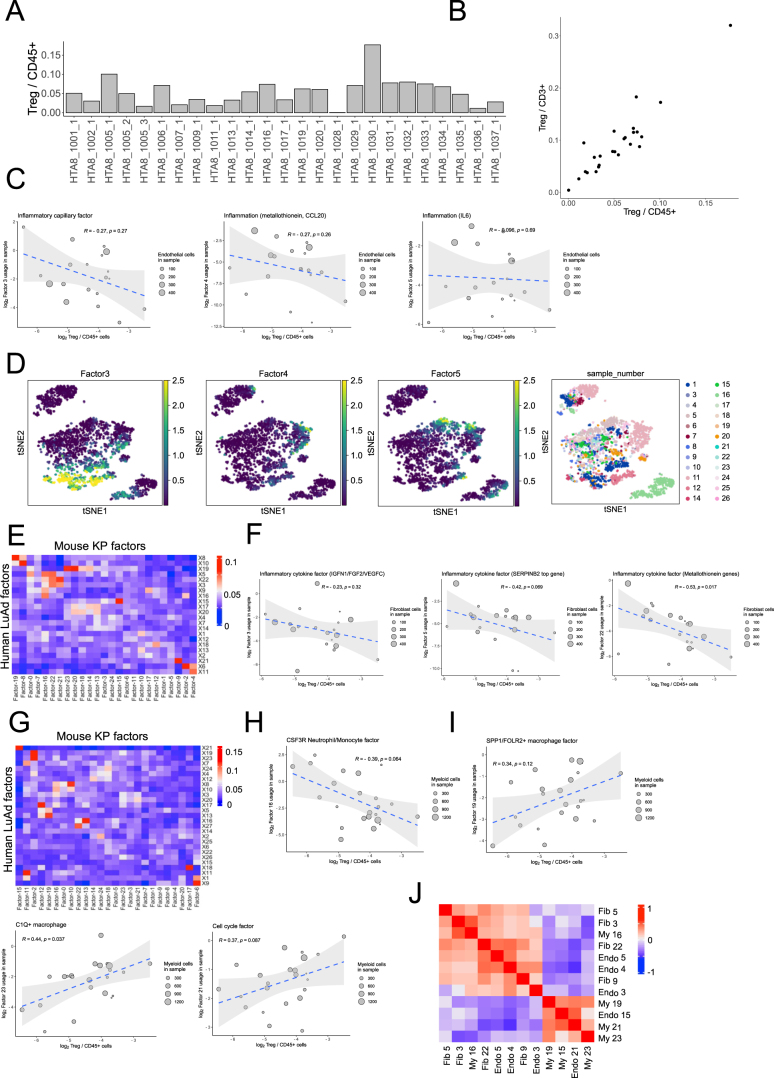
While regulatory T (Treg) cells are traditionally viewed as professional suppressors of antigen presenting cells and effector T cells in both autoimmunity and cancer, recent findings of distinct Treg cell functions in tissue maintenance suggest that their regulatory purview extends to a wider range of cells and is broader than previously assumed. To elucidate tumoral Treg cell 'connectivity' to diverse tumor-supporting accessory cell types, we explored immediate early changes in their single-cell transcriptomes upon punctual Treg cell depletion in experimental lung cancer and injury-induced inflammation. Before any notable T cell activation and inflammation, fibroblasts, endothelial and myeloid cells exhibited pronounced changes in their gene expression in both cancer and injury settings. Factor analysis revealed shared Treg cell-dependent gene programs, foremost, prominent upregulation of VEGF and CCR2 signaling-related genes upon Treg cell deprivation in either setting, as well as in Treg cell-poor versus Treg cell-rich human lung adenocarcinomas. Accordingly, punctual Treg cell depletion combined with short-term VEGF blockade showed markedly improved control of PD-1 blockade-resistant lung adenocarcinoma progression in mice compared to the corresponding monotherapies, highlighting a promising factor-based querying approach to elucidating new rational combination treatments of solid organ cancers.
© 2023. The Author(s).
Conflict of interest statement
A.Y.R. is a member of SAB, and has equity in Surface Oncology, RAPT Therapeutics, Sonoma Biotherapeutics, Santa Ana Bio and Vedanta Biosciences and is an SAB member of BioInvent and Amgen; A.Y.R. holds a therapeutic Treg cell depletion IP licensed to Takeda. C.M.R. has consulted regarding oncology drug development with AbbVie, Amgen, Astra Zeneca, D2G, Daiichi Sankyo, Epizyme, Genentech/Roche, Ipsen, Jazz, Kowa, and Merck, and is a member of the SAB of Auron, Bridge Medicines, Earli, and Harpoon Therapeutics. D.P is a member of the SAB and has equity in Insitro. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
