

β-Catenin-Driven Differentiation Is a Tissue-Specific Epigenetic Vulnerability in Adrenal Cancer
- PMID: 37129912
- PMCID: PMC10330305
- DOI: 10.1158/0008-5472.CAN-22-2712
β-Catenin-Driven Differentiation Is a Tissue-Specific Epigenetic Vulnerability in Adrenal Cancer
Abstract
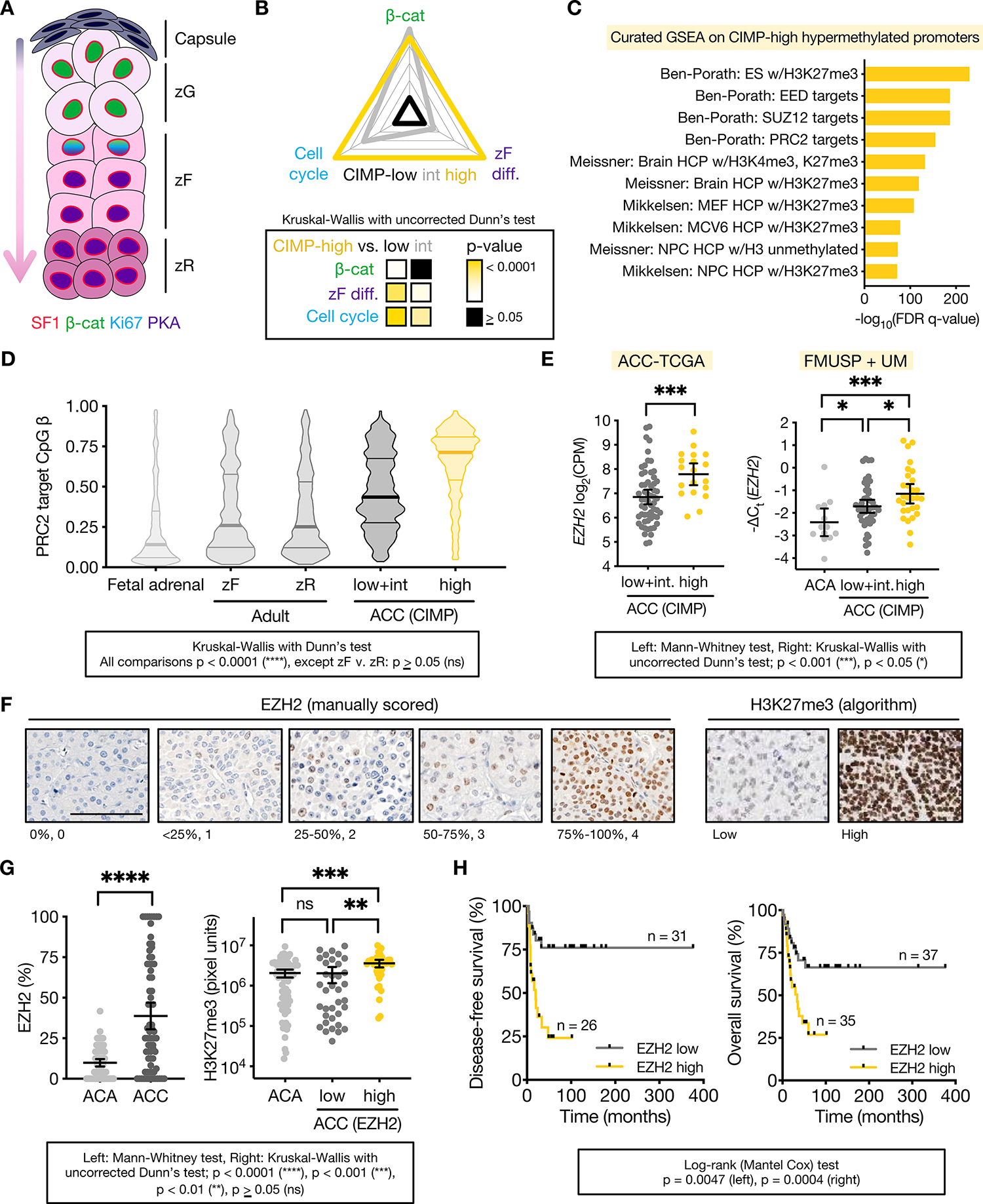
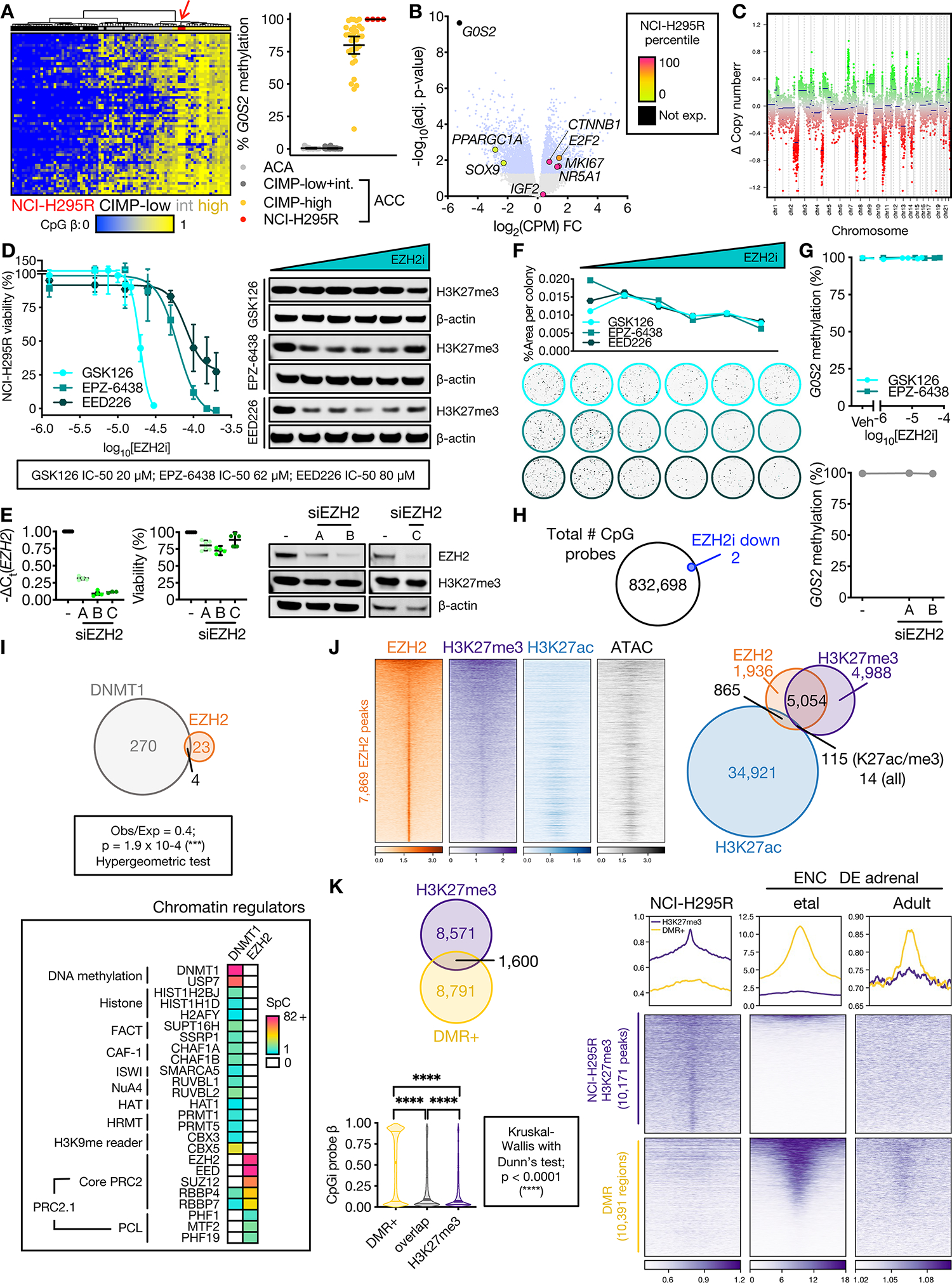
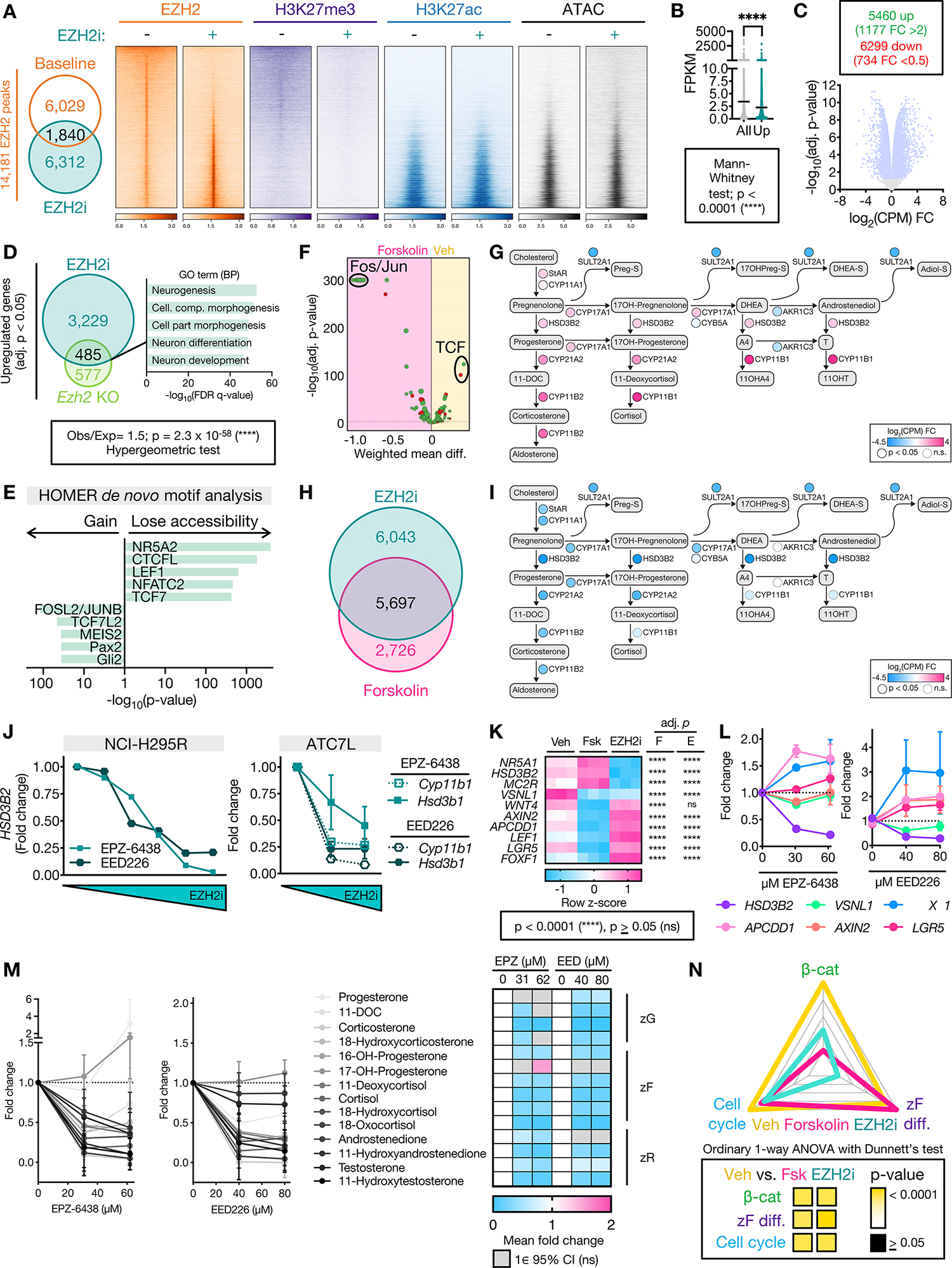
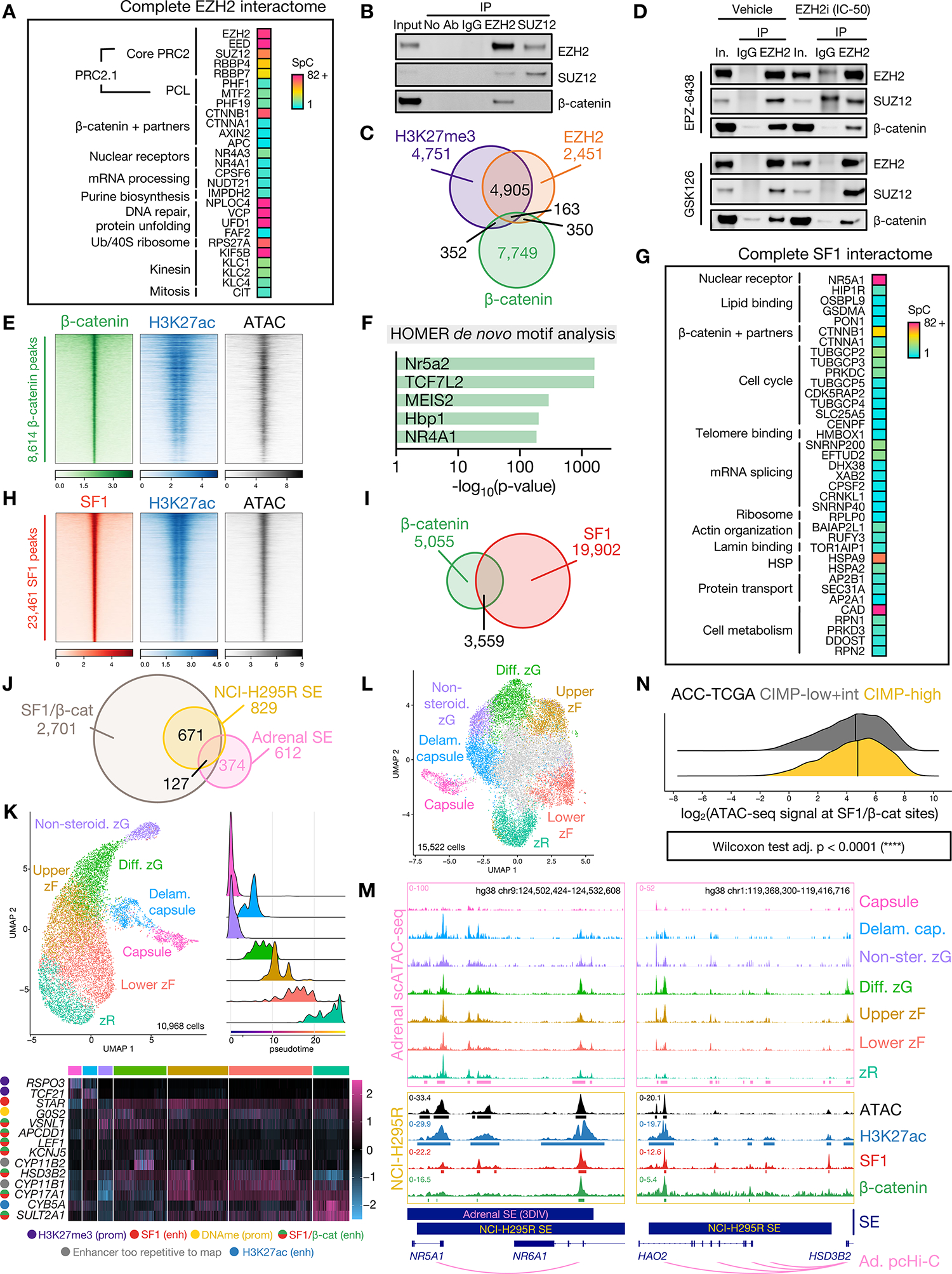
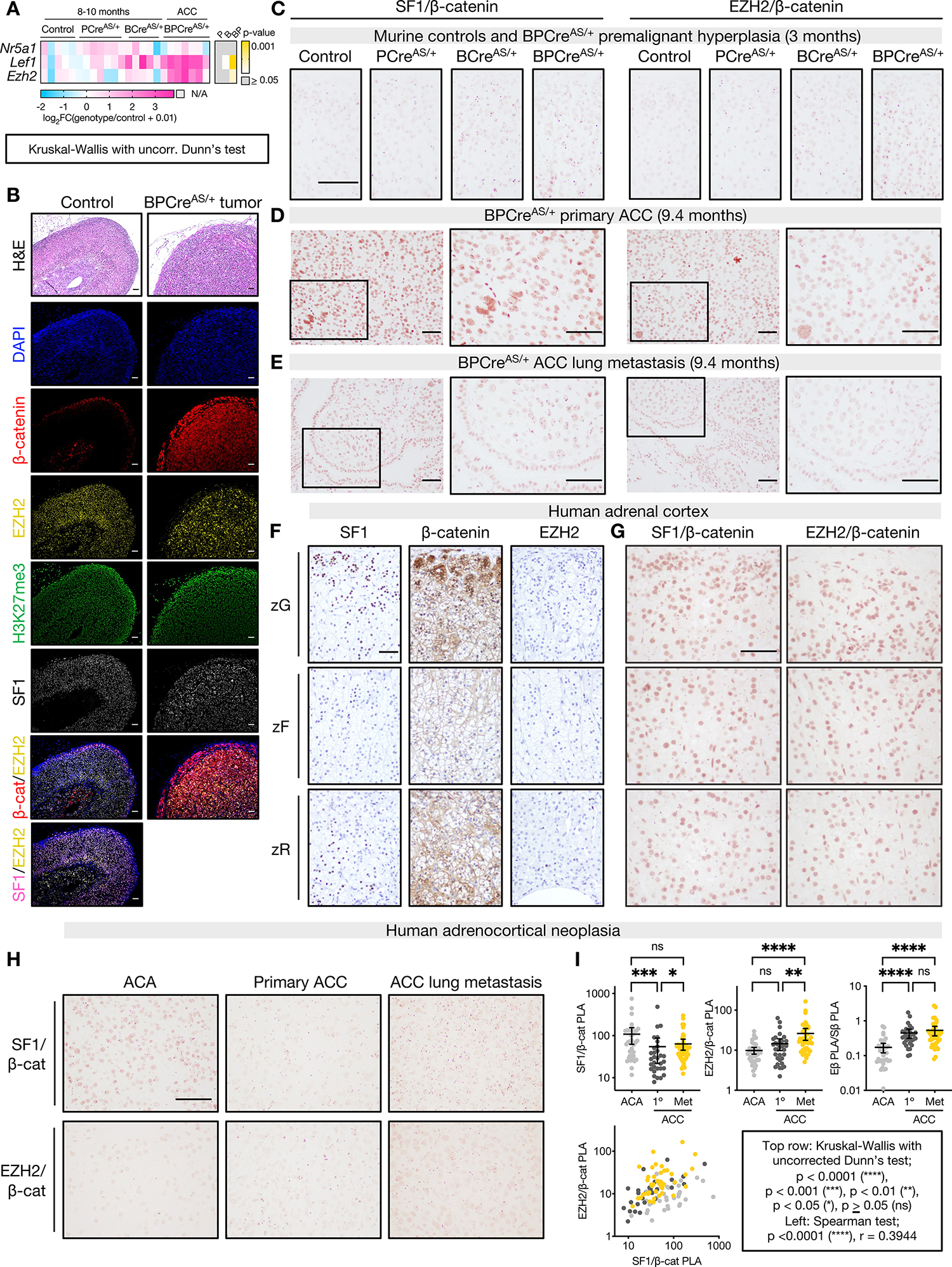
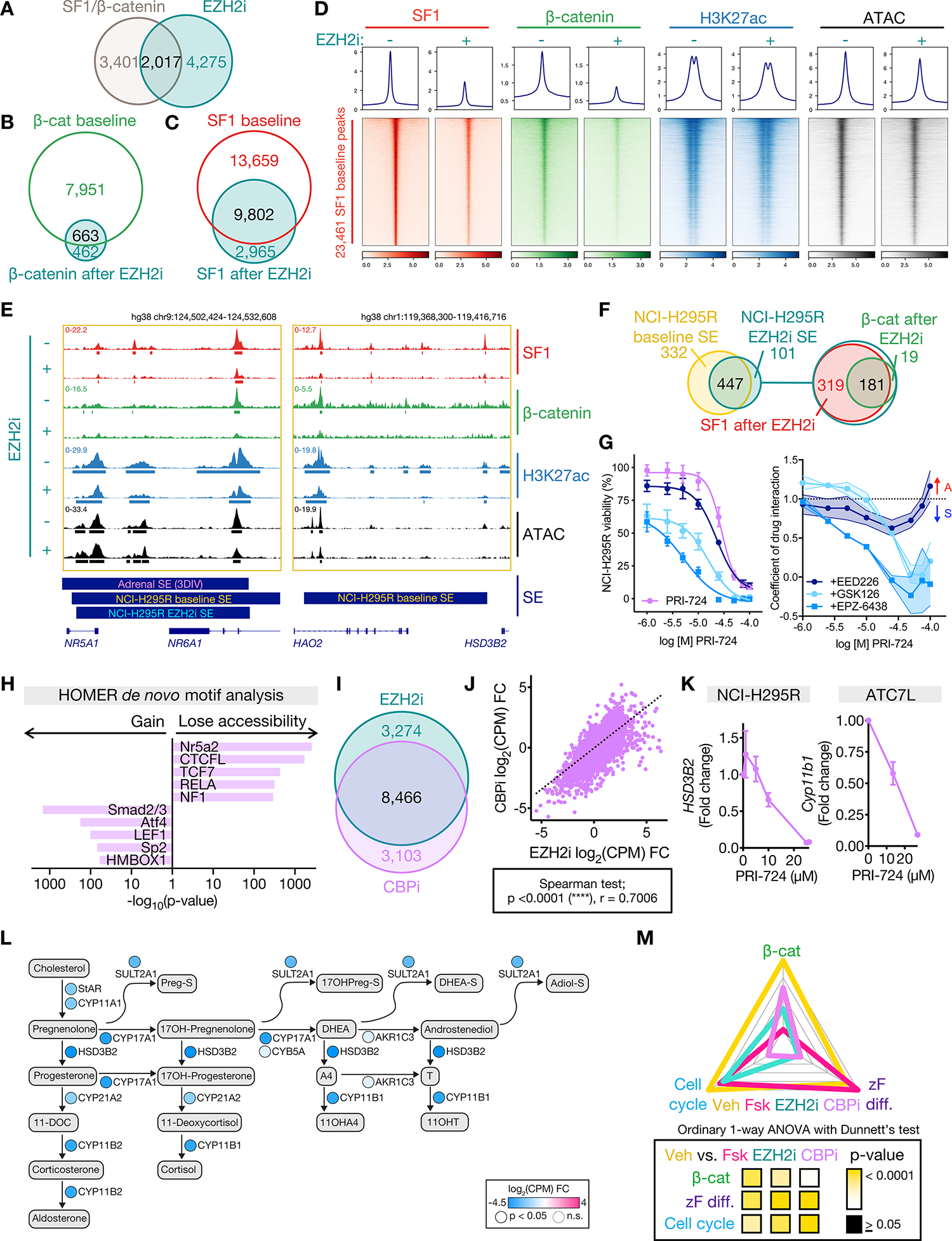
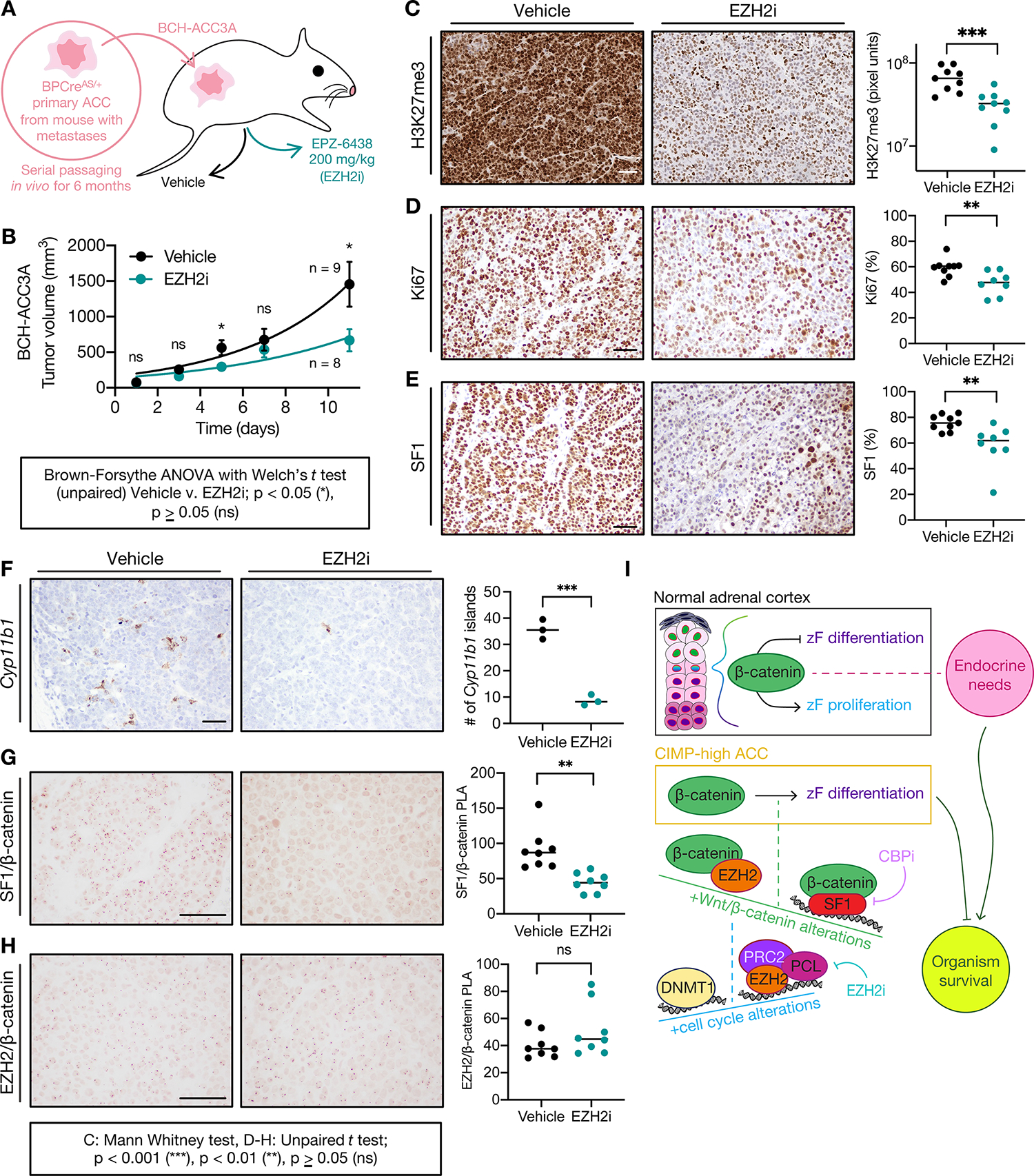
Adrenocortical carcinoma (ACC) is a rare cancer in which tissue-specific differentiation is paradoxically associated with dismal outcomes. The differentiated ACC subtype CIMP-high is prevalent, incurable, and routinely fatal. CIMP-high ACC possess abnormal DNA methylation and frequent β-catenin-activating mutations. Here, we demonstrated that ACC differentiation is maintained by a balance between nuclear, tissue-specific β-catenin-containing complexes, and the epigenome. On chromatin, β-catenin bound master adrenal transcription factor SF1 and hijacked the adrenocortical super-enhancer landscape to maintain differentiation in CIMP-high ACC; off chromatin, β-catenin bound histone methyltransferase EZH2. SF1/β-catenin and EZH2/β-catenin complexes present in normal adrenals persisted through all phases of ACC evolution. Pharmacologic EZH2 inhibition in CIMP-high ACC expelled SF1/β-catenin from chromatin and favored EZH2/β-catenin assembly, erasing differentiation and restraining cancer growth in vitro and in vivo. These studies illustrate how tissue-specific programs shape oncogene selection, surreptitiously encoding targetable therapeutic vulnerabilities.
Significance: Oncogenic β-catenin can use tissue-specific partners to regulate cellular differentiation programs that can be reversed by epigenetic therapies, identifying epigenetic control of differentiation as a viable target for β-catenin-driven cancers.
©2023 American Association for Cancer Research.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
D.R. Mohan, A.M. Lerario, and G.D. Hammer are inventors on three pending patent applications describing compositions and methods for treating or characterizing cancer. G.D. Hammer reports unrelated personal fees from Radionetics and Orphagen Pharmaceuticals for consultation on projects outside the scope of this work. The remaining authors declare no competing interests.
Figures
References
-
- Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017;171:34–57 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
