

Algorithm for optimized mRNA design improves stability and immunogenicity
- PMID: 37130545
- PMCID: PMC10499610
- DOI: 10.1038/s41586-023-06127-z
Algorithm for optimized mRNA design improves stability and immunogenicity
Abstract
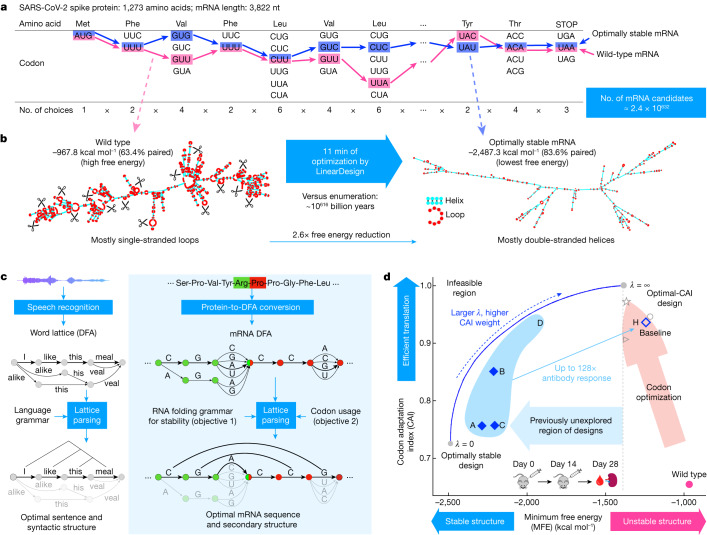
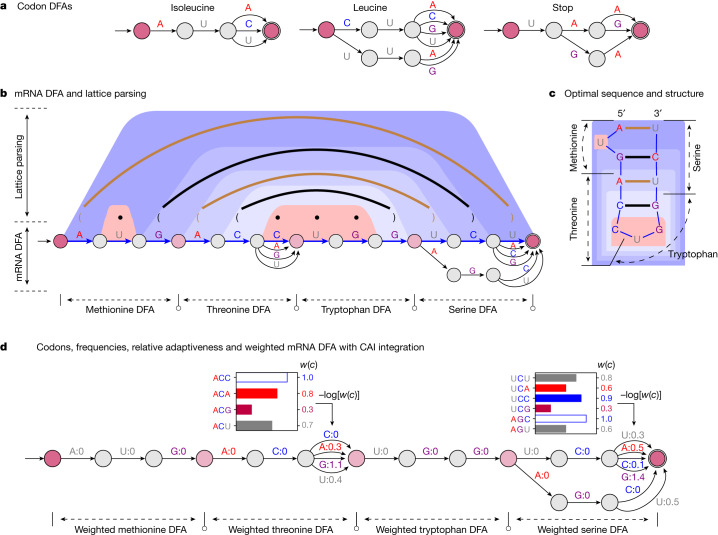
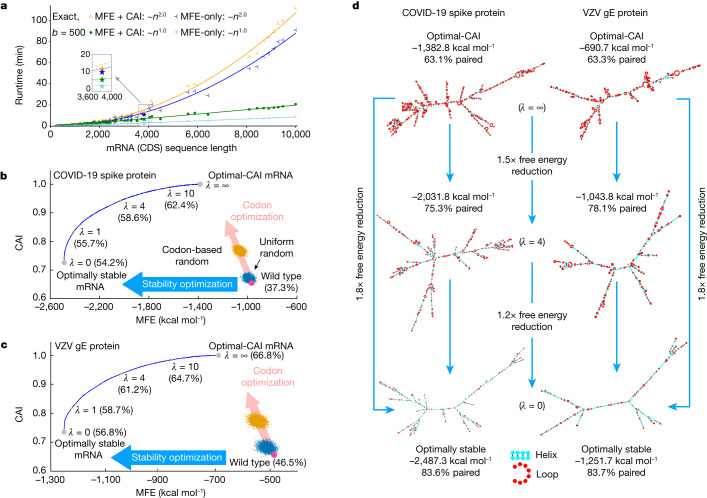
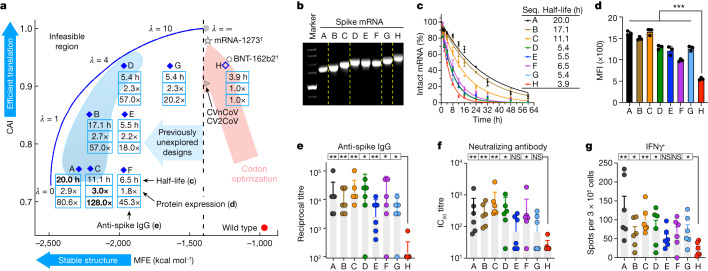
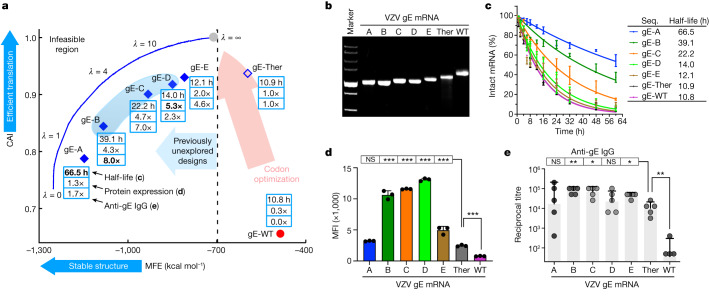
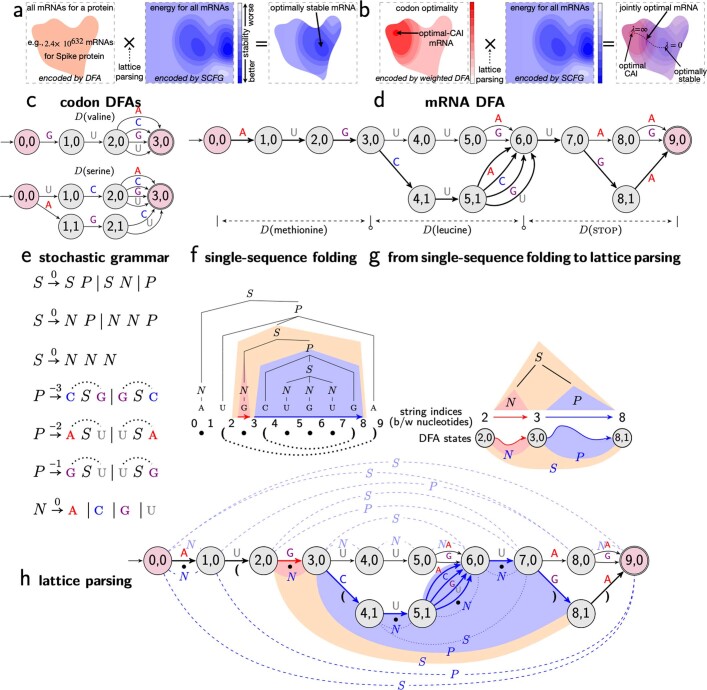
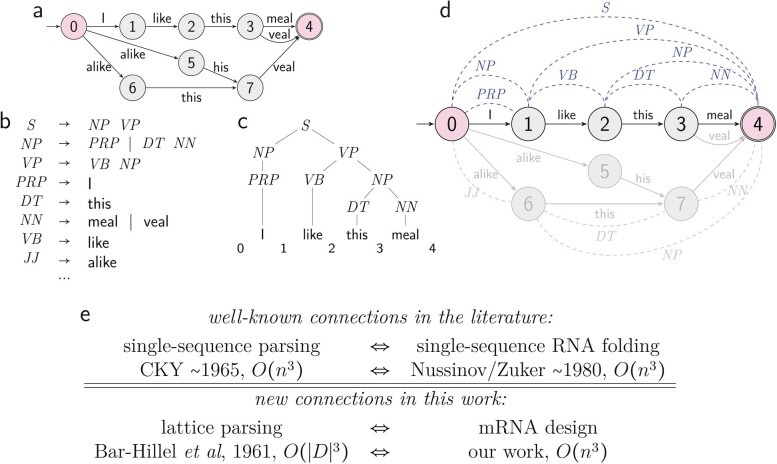
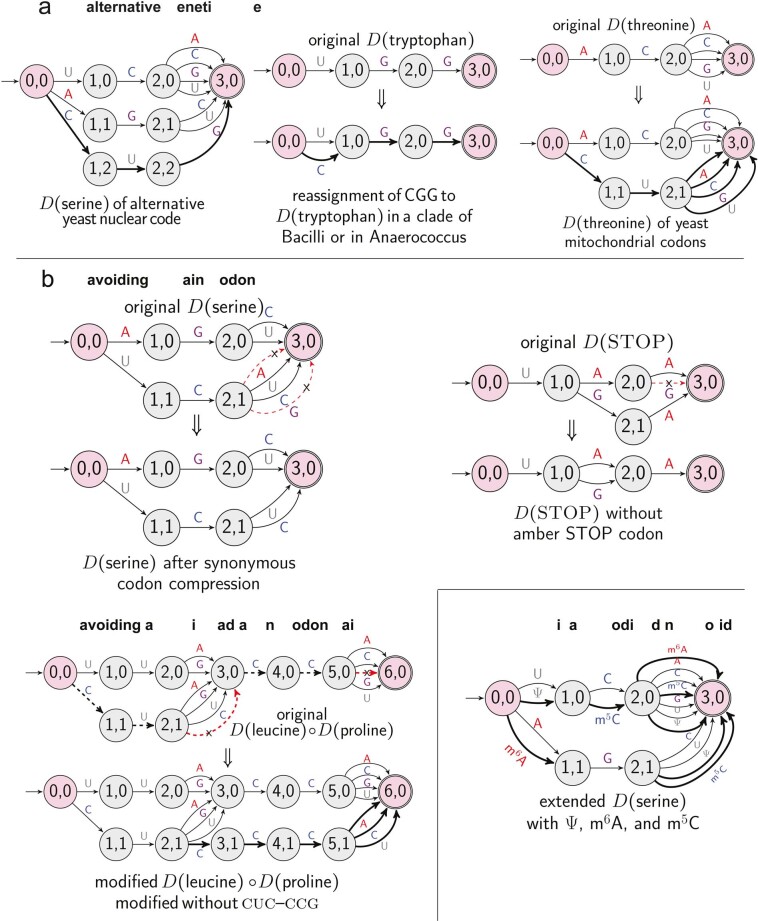
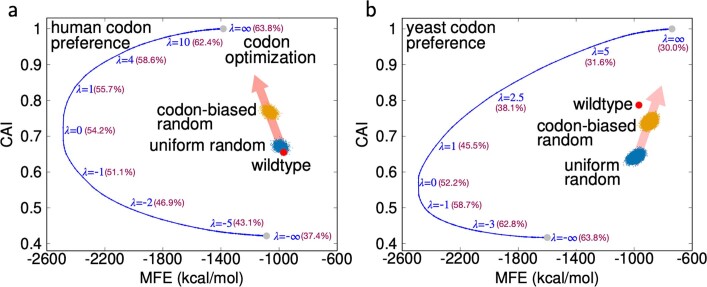
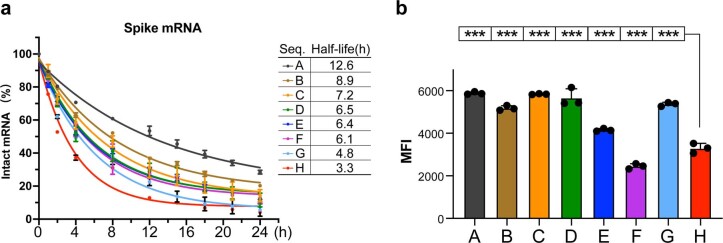
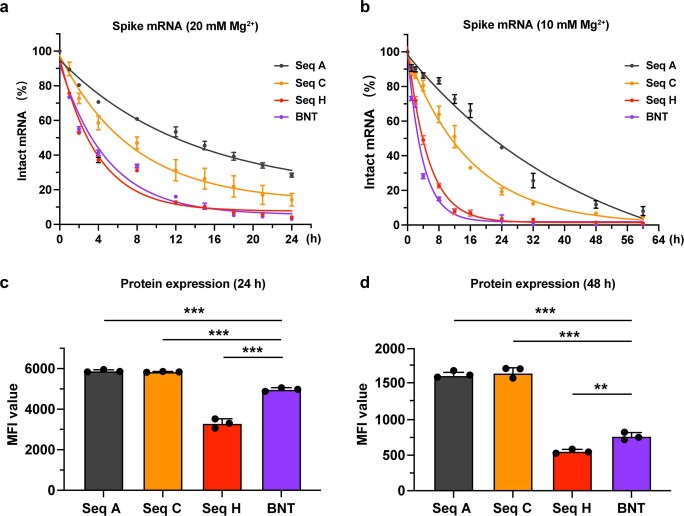
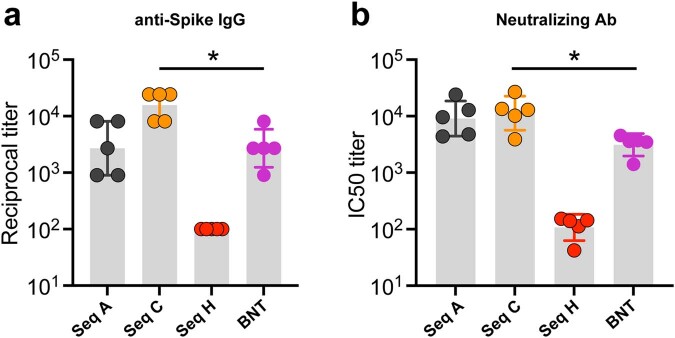
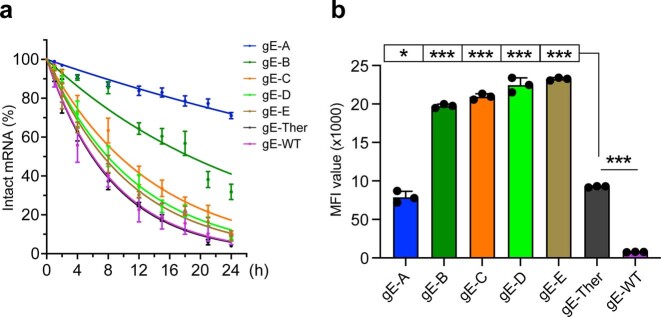
Messenger RNA (mRNA) vaccines are being used to combat the spread of COVID-19 (refs. 1-3), but they still exhibit critical limitations caused by mRNA instability and degradation, which are major obstacles for the storage, distribution and efficacy of the vaccine products4. Increasing secondary structure lengthens mRNA half-life, which, together with optimal codons, improves protein expression5. Therefore, a principled mRNA design algorithm must optimize both structural stability and codon usage. However, owing to synonymous codons, the mRNA design space is prohibitively large-for example, there are around 2.4 × 10632 candidate mRNA sequences for the SARS-CoV-2 spike protein. This poses insurmountable computational challenges. Here we provide a simple and unexpected solution using the classical concept of lattice parsing in computational linguistics, where finding the optimal mRNA sequence is analogous to identifying the most likely sentence among similar-sounding alternatives6. Our algorithm LinearDesign finds an optimal mRNA design for the spike protein in just 11 minutes, and can concurrently optimize stability and codon usage. LinearDesign substantially improves mRNA half-life and protein expression, and profoundly increases antibody titre by up to 128 times in mice compared to the codon-optimization benchmark on mRNA vaccines for COVID-19 and varicella-zoster virus. This result reveals the great potential of principled mRNA design and enables the exploration of previously unreachable but highly stable and efficient designs. Our work is a timely tool for vaccines and other mRNA-based medicines encoding therapeutic proteins such as monoclonal antibodies and anti-cancer drugs7,8.
© 2023. The Author(s).
Conflict of interest statement
Baidu USA filed a patent for the LinearDesign algorithm in 2021 listing H.Z., L.Z., Z.L., K.L., B.L. and L.H. as inventors. The work of H.Z., L.Z., Z.L., K.L., B.L. and L.H. for the development of the LinearDesign algorithm was conducted at Baidu USA. StemiRNA Therapeutics has filed a provisional patent for the VZV mRNA vaccine listing C.X., H.S. and H.L. as inventors. Sanofi entered a non-exclusive licensing agreement with Baidu USA in 2021 to use LinearDesign to develop mRNA vaccines and therapeutics. H.Z., L.Z., Z.L., K.L., B.L. and L.H. are or were employees of Baidu USA. A.L., C.X., X.M., F.Z., H.J., C.C., H.S., H.L. and Y.Z. are or were employees of StemiRNA Therapeutics. L.H. and D.H.M. are also cofounders of Coderna.ai.
Figures
Comment in
-
A tool for optimizing messenger RNA sequence.Nature. 2023 Sep;621(7978):262-264. doi: 10.1038/d41586-023-01745-z. Nature. 2023. PMID: 37253980 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
