

This is a preprint.
DNA damage-induced interaction between a lineage addiction oncogenic transcription factor and the MRN complex shapes a tissue-specific DNA Damage Response and cancer predisposition
- PMID: 37131595
- PMCID: PMC10153263
- DOI: 10.1101/2023.04.21.537819
DNA damage-induced interaction between a lineage addiction oncogenic transcription factor and the MRN complex shapes a tissue-specific DNA Damage Response and cancer predisposition
Abstract
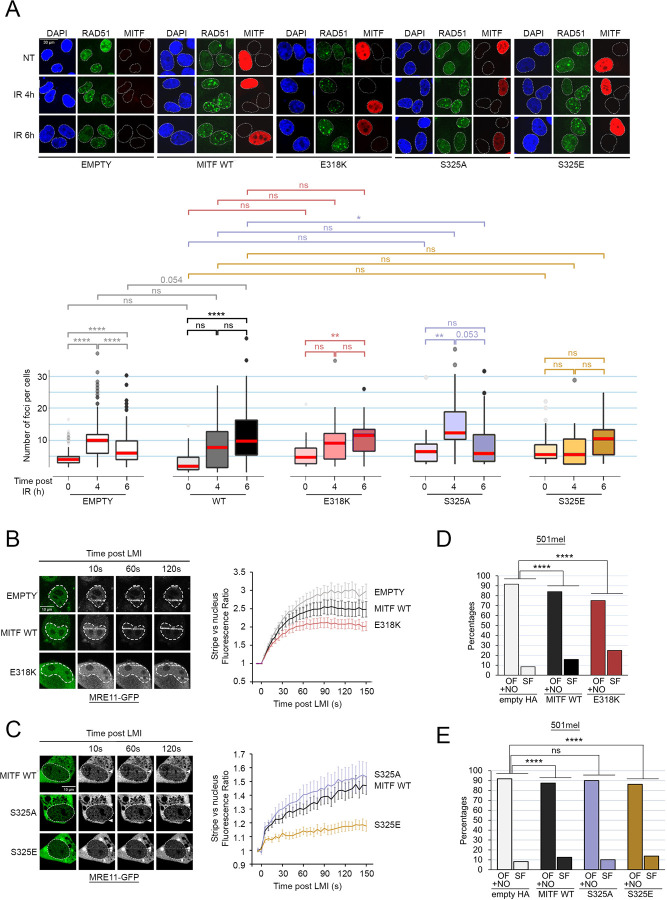
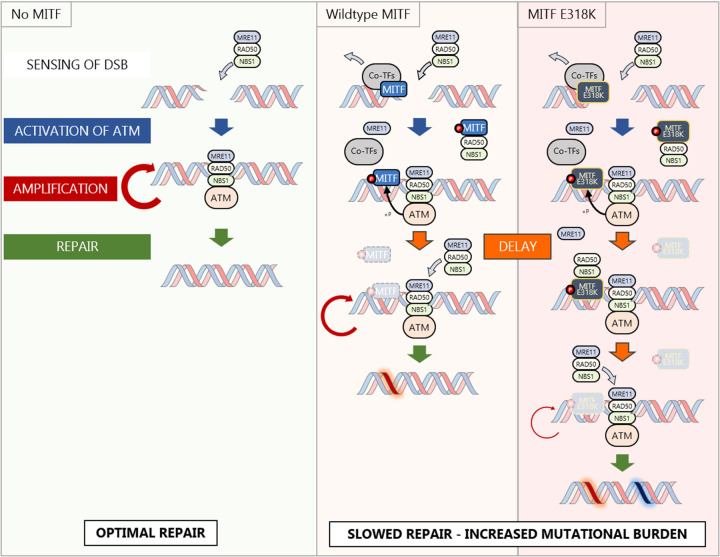
Since genome instability can drive cancer initiation and progression, cells have evolved highly effective and ubiquitous DNA Damage Response (DDR) programs. However, some cells, in skin for example, are normally exposed to high levels of DNA damaging agents. Whether such high-risk cells possess lineage-specific mechanisms that tailor DNA repair to the tissue remains largely unknown. Here we show, using melanoma as a model, that the microphthalmia-associated transcription factor MITF, a lineage addition oncogene that coordinates many aspects of melanocyte and melanoma biology, plays a non-transcriptional role in shaping the DDR. On exposure to DNA damaging agents, MITF is phosphorylated by ATM/DNA-PKcs, and unexpectedly its interactome is dramatically remodelled; most transcription (co)factors dissociate, and instead MITF interacts with the MRE11-RAD50-NBS1 (MRN) complex. Consequently, cells with high MITF levels accumulate stalled replication forks, and display defects in homologous recombination-mediated repair associated with impaired MRN recruitment to DNA damage. In agreement, high MITF levels are associated with increased SNV burden in melanoma. Significantly, the SUMOylation-defective MITF-E318K melanoma predisposition mutation recapitulates the effects of ATM/DNA-PKcs-phosphorylated MITF. Our data suggest that a non-transcriptional function of a lineage-restricted transcription factor contributes to a tissue-specialised modulation of the DDR that can impact cancer initiation.
Keywords: DNA Damage repair; DNA Replication; E318K; Homologous Recombination; MITF; Melanoma; NBS1; Replication Stress; SUMOylation.
Conflict of interest statement
Competing Interest Statement The authors declare no competing interests.
Figures




References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous