

Cytosolic calcium: Judge, jury and executioner of neurodegeneration in Alzheimer's disease and beyond
- PMID: 37132525
- PMCID: PMC10490830
- DOI: 10.1002/alz.13065
Cytosolic calcium: Judge, jury and executioner of neurodegeneration in Alzheimer's disease and beyond
Abstract
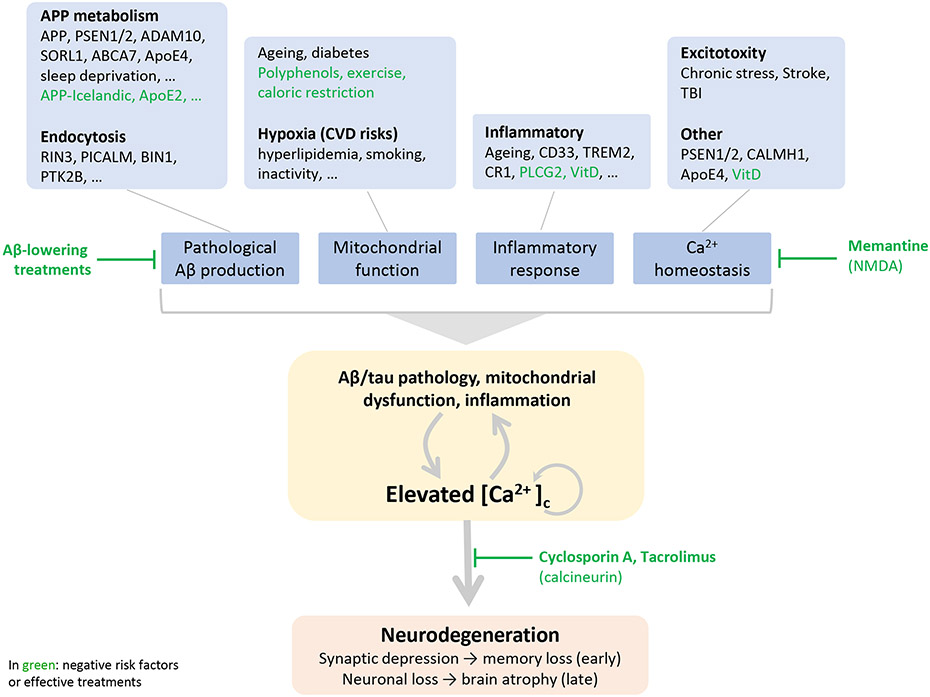
This review discusses the driving principles that may underlie neurodegeneration in dementia, represented most dominantly by Alzheimer's disease (AD). While a myriad of different disease risk factors contribute to AD, these ultimately converge to a common disease outcome. Based on decades of research, a picture emerges where upstream risk factors combine in a feedforward pathophysiological cycle, culminating in a rise of cytosolic calcium concentration ([Ca2+ ]c ) that triggers neurodegeneration. In this framework, positive AD risk factors entail conditions, characteristics, or lifestyles that initiate or accelerate self-reinforcing cycles of pathophysiology, whereas negative risk factors or therapeutic interventions, particularly those mitigating elevated [Ca2+ ]c , oppose these effects and therefore have neuroprotective potential.
Keywords: Alzheimer's disease; calcineurin; calcium; dementia; neurodegeneration; synaptic function.
© 2023 reMYND N.V and The Authors. Alzheimer's & Dementia published by Wiley Periodicals LLC on behalf of Alzheimer's Association.
Conflict of interest statement
Conflicts of interests
GG is consultant for reMYND. MF is full time employee of reMYND. GG and MF own reMYND warrants and/or stocks. EKW and GES have nothing to disclose.
Figures





References
-
- Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 1991;30(4):572–580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
