

The electron-proton bottleneck of photosynthetic oxygen evolution
- PMID: 37138082
- PMCID: PMC10191853
- DOI: 10.1038/s41586-023-06008-5
The electron-proton bottleneck of photosynthetic oxygen evolution
Abstract
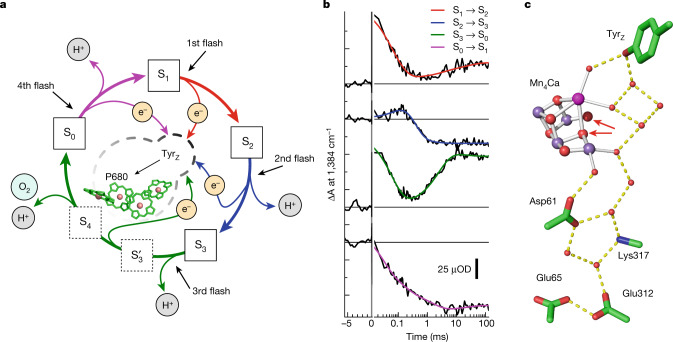
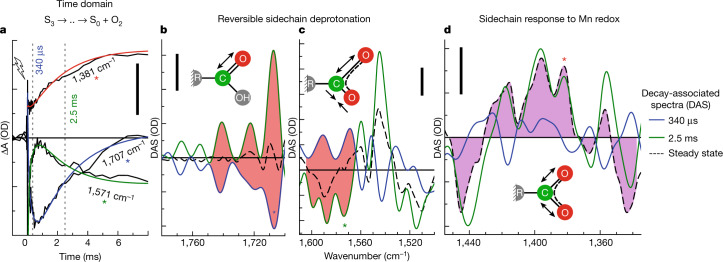
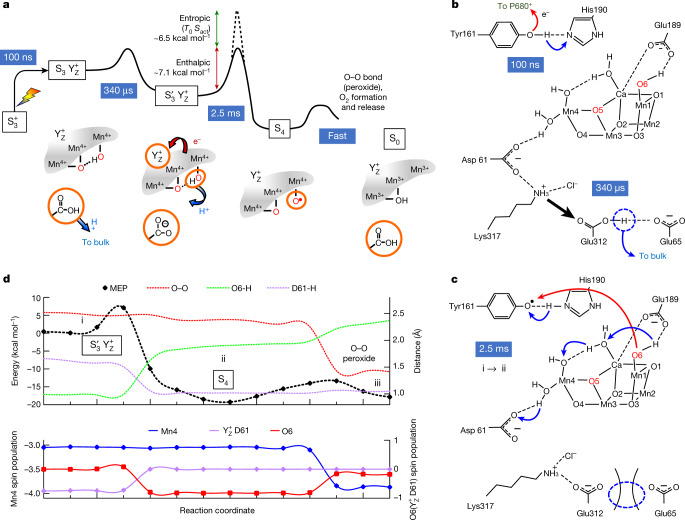
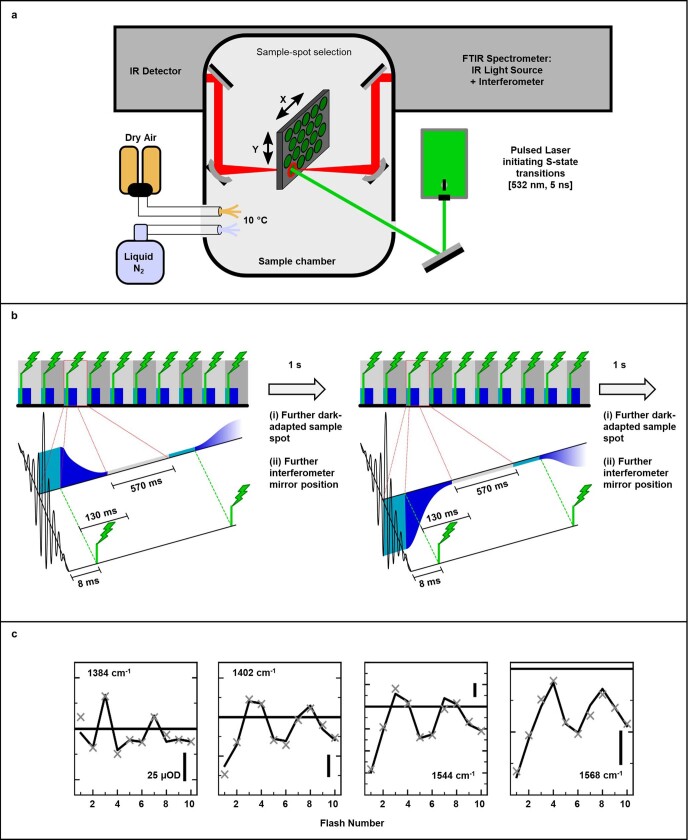
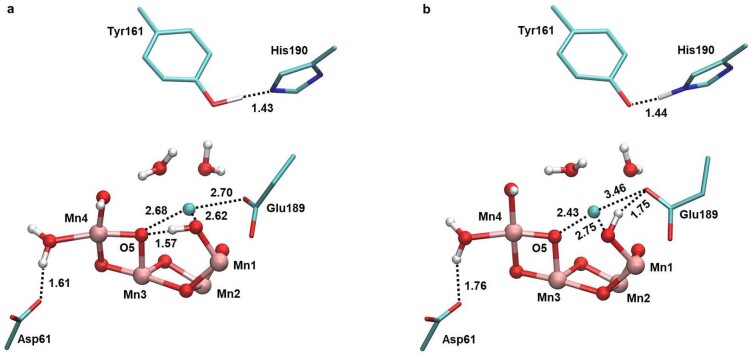
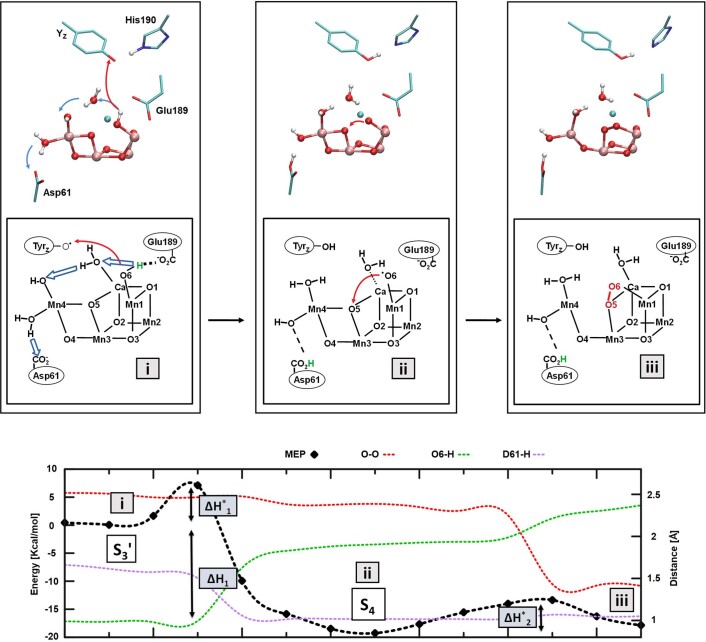
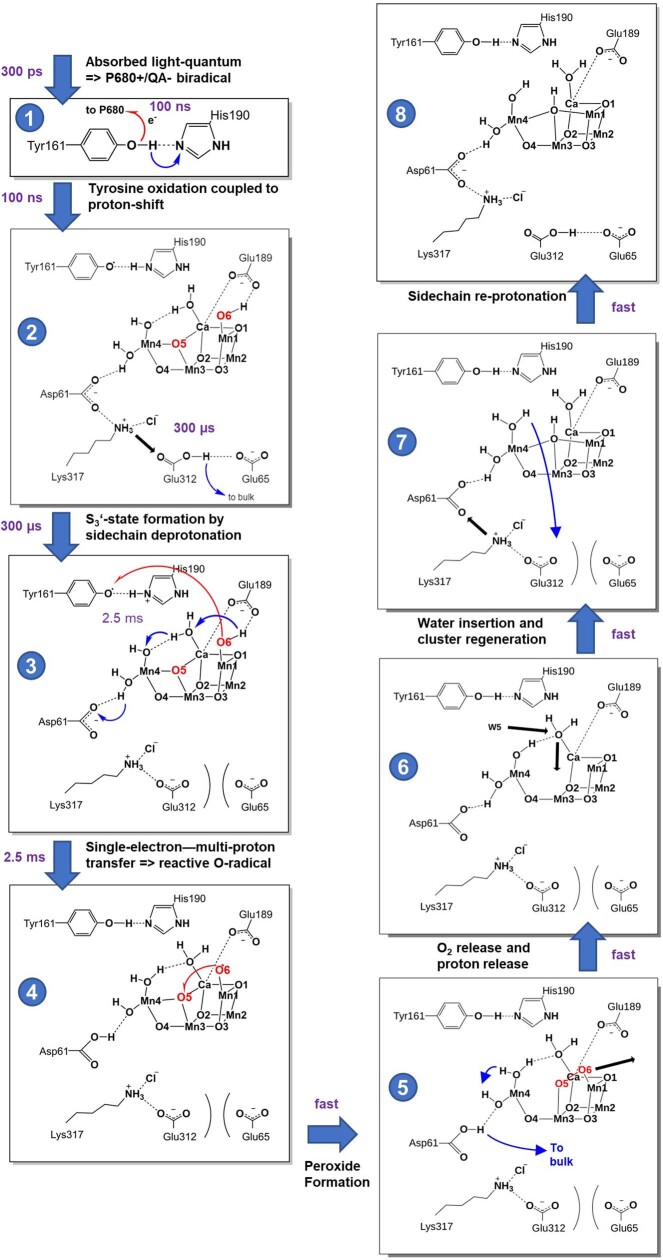
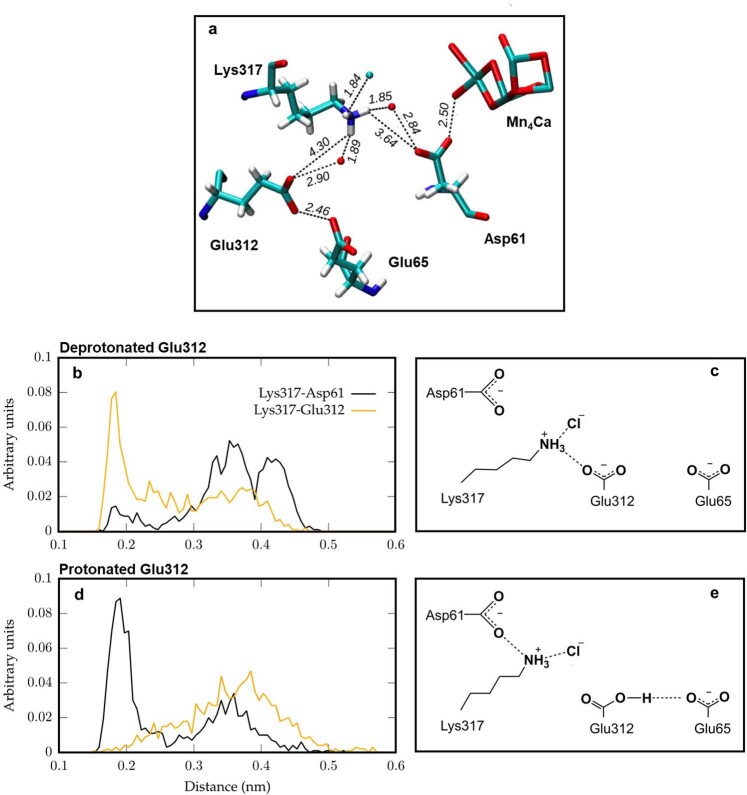
Photosynthesis fuels life on Earth by storing solar energy in chemical form. Today's oxygen-rich atmosphere has resulted from the splitting of water at the protein-bound manganese cluster of photosystem II during photosynthesis. Formation of molecular oxygen starts from a state with four accumulated electron holes, the S4 state-which was postulated half a century ago1 and remains largely uncharacterized. Here we resolve this key stage of photosynthetic O2 formation and its crucial mechanistic role. We tracked 230,000 excitation cycles of dark-adapted photosystems with microsecond infrared spectroscopy. Combining these results with computational chemistry reveals that a crucial proton vacancy is initally created through gated sidechain deprotonation. Subsequently, a reactive oxygen radical is formed in a single-electron, multi-proton transfer event. This is the slowest step in photosynthetic O2 formation, with a moderate energetic barrier and marked entropic slowdown. We identify the S4 state as the oxygen-radical state; its formation is followed by fast O-O bonding and O2 release. In conjunction with previous breakthroughs in experimental and computational investigations, a compelling atomistic picture of photosynthetic O2 formation emerges. Our results provide insights into a biological process that is likely to have occurred unchanged for the past three billion years, which we expect to support the knowledge-based design of artificial water-splitting systems.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
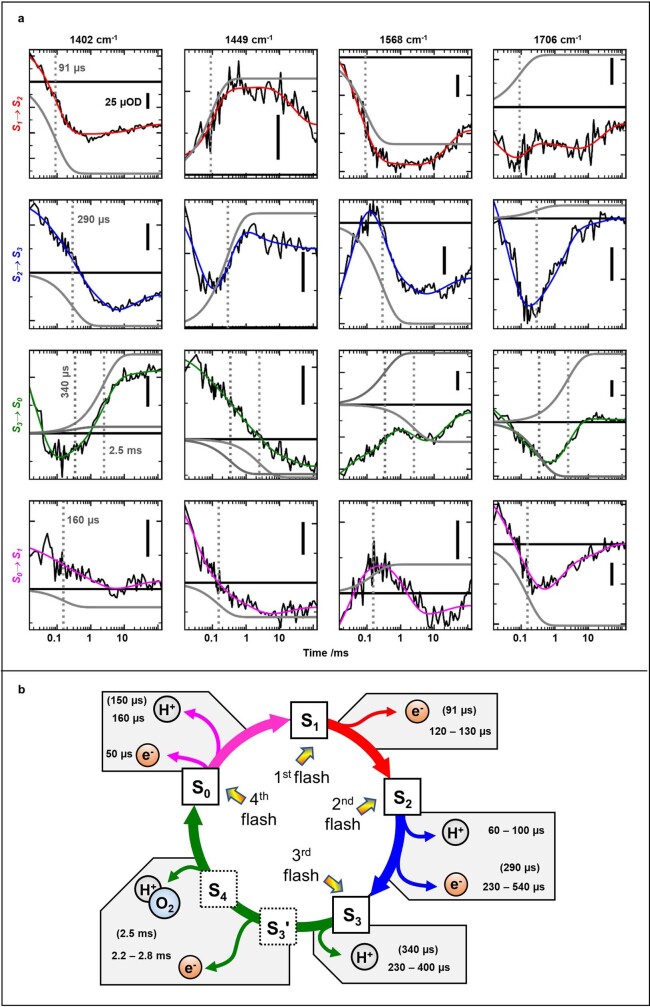
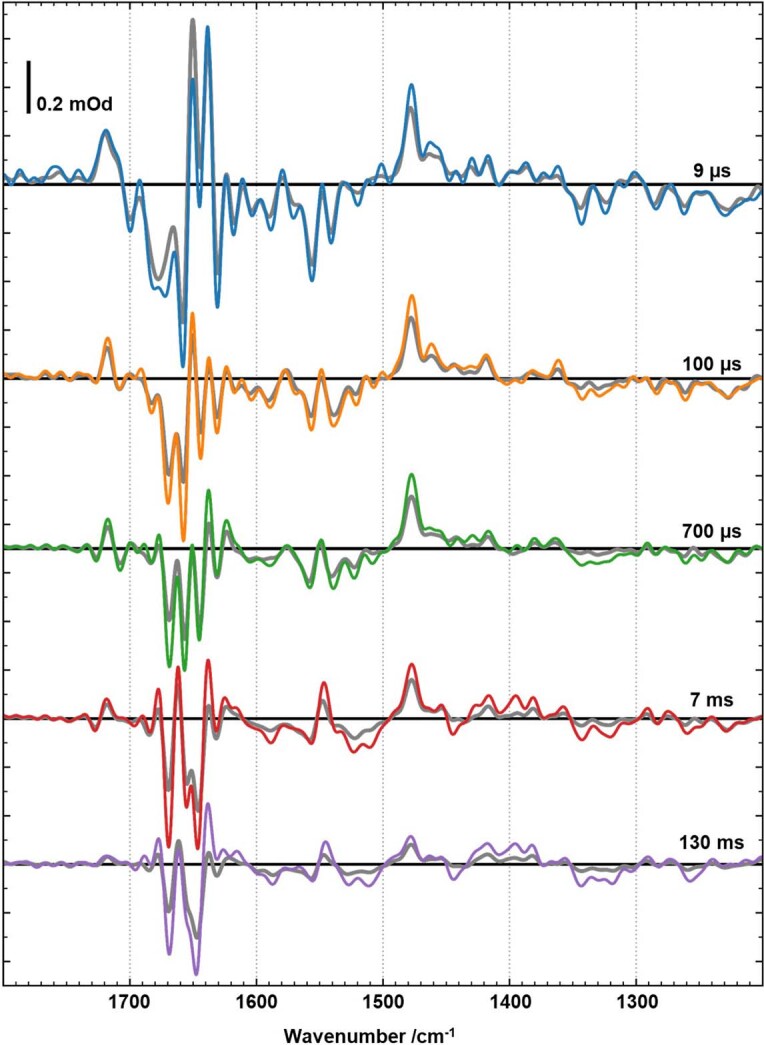
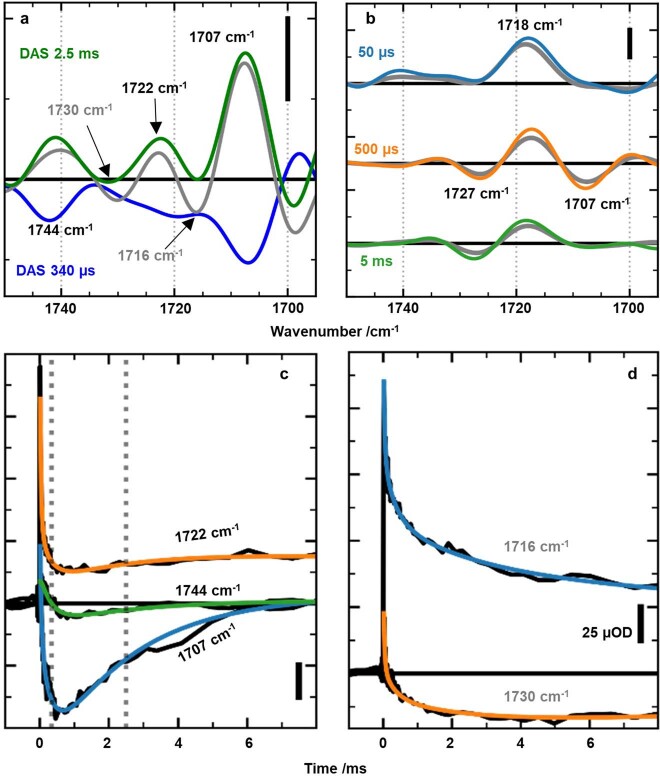
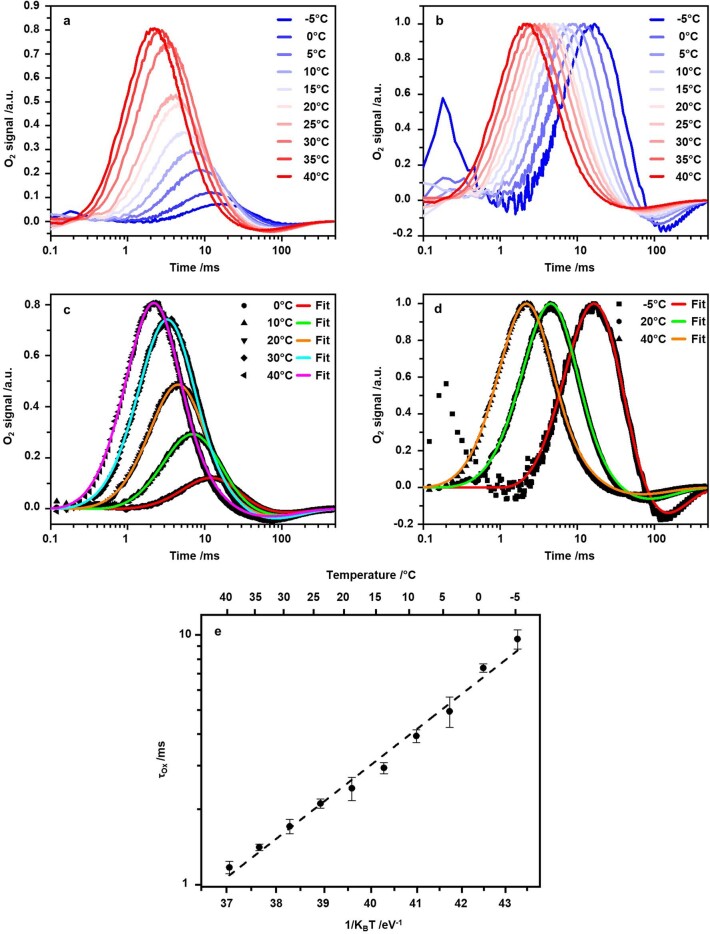
Comment in
-
Clues to how water splits during photosynthesis.Nature. 2023 May;617(7961):468-469. doi: 10.1038/d41586-023-01388-0. Nature. 2023. PMID: 37138059 No abstract available.
Similar articles
-
Mechanism of proton release during water oxidation in Photosystem II.Proc Natl Acad Sci U S A. 2024 Dec 24;121(52):e2413396121. doi: 10.1073/pnas.2413396121. Epub 2024 Dec 19. Proc Natl Acad Sci U S A. 2024. PMID: 39700151 Free PMC article.
-
Eight steps preceding O-O bond formation in oxygenic photosynthesis--a basic reaction cycle of the Photosystem II manganese complex.Biochim Biophys Acta. 2007 Jun;1767(6):472-83. doi: 10.1016/j.bbabio.2007.02.022. Epub 2007 Mar 12. Biochim Biophys Acta. 2007. PMID: 17442260 Review.
-
Photosynthetic O2 formation tracked by time-resolved x-ray experiments.Science. 2005 Nov 11;310(5750):1019-21. doi: 10.1126/science.1117551. Science. 2005. PMID: 16284178
-
Structural evidence for intermediates during O2 formation in photosystem II.Nature. 2023 May;617(7961):629-636. doi: 10.1038/s41586-023-06038-z. Epub 2023 May 3. Nature. 2023. PMID: 37138085 Free PMC article.
-
Fourier transform infrared difference and time-resolved infrared detection of the electron and proton transfer dynamics in photosynthetic water oxidation.Biochim Biophys Acta. 2015 Jan;1847(1):35-45. doi: 10.1016/j.bbabio.2014.06.009. Epub 2014 Jul 3. Biochim Biophys Acta. 2015. PMID: 24998309 Review.
Cited by
-
Nature of S-States in the Oxygen-Evolving Complex Resolved by High-Energy Resolution Fluorescence Detected X-ray Absorption Spectroscopy.J Am Chem Soc. 2023 Nov 29;145(47):25579-25594. doi: 10.1021/jacs.3c06046. Epub 2023 Nov 16. J Am Chem Soc. 2023. PMID: 37970825 Free PMC article.
-
Mutation-induced shift of the photosystem II active site reveals insight into conserved water channels.J Biol Chem. 2024 Jul;300(7):107475. doi: 10.1016/j.jbc.2024.107475. Epub 2024 Jun 13. J Biol Chem. 2024. PMID: 38879008 Free PMC article.
-
Unravelling Mn4Ca cluster vibrations in the S1, S2 and S3 states of the Kok-Joliot cycle of photosystem II.Phys Chem Chem Phys. 2024 Jul 31;26(30):20598-20609. doi: 10.1039/d4cp01307g. Phys Chem Chem Phys. 2024. PMID: 39037338 Free PMC article.
-
The S1 to S2 and S2 to S3 state transitions in plant photosystem II: relevance to the functional and structural heterogeneity of the water oxidizing complex.Photosynth Res. 2024 Dec;162(2-3):401-411. doi: 10.1007/s11120-024-01096-4. Epub 2024 Apr 25. Photosynth Res. 2024. PMID: 38662327 Free PMC article.
-
Mechanism of proton release during water oxidation in Photosystem II.Proc Natl Acad Sci U S A. 2024 Dec 24;121(52):e2413396121. doi: 10.1073/pnas.2413396121. Epub 2024 Dec 19. Proc Natl Acad Sci U S A. 2024. PMID: 39700151 Free PMC article.
References
-
- Kok B, Forbush B, McGloin M. Cooperation of charges in photosynthetic O2 evolution - I. A linear four-step mechanism. Photochem. Photobiol. 1970;11:457–475. - PubMed
-
- Junge W. Oxygenic photosynthesis: history, status and perspective. Q. Rev. Biophys. 2019;52:e1. - PubMed
-
- Fischer WW, Hemp J, Johnson JE. Manganese and the evolution of photosynthesis. Orig. Life Evol. Biosph. 2015;45:351–357. - PubMed
-
- Cox N, Pantazis DA, Lubitz W. Current understanding of the mechanism of water oxidation in photosystem II and its relation to XFEL data. Annu. Rev. Biochem. 2020;89:795–820. - PubMed
-
- Noguchi T, Suzuki H, Tsuno M, Sugiura M, Kato C. Time-resolved infrared detection of the proton and protein dynamics during photosynthetic oxygen evolution. Biochemistry. 2012;51:3205–3214. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
