

Re-convolving the compositional landscape of primary and recurrent glioblastoma reveals prognostic and targetable tissue states
- PMID: 37142563
- PMCID: PMC10160047
- DOI: 10.1038/s41467-023-38186-1
Re-convolving the compositional landscape of primary and recurrent glioblastoma reveals prognostic and targetable tissue states
Abstract
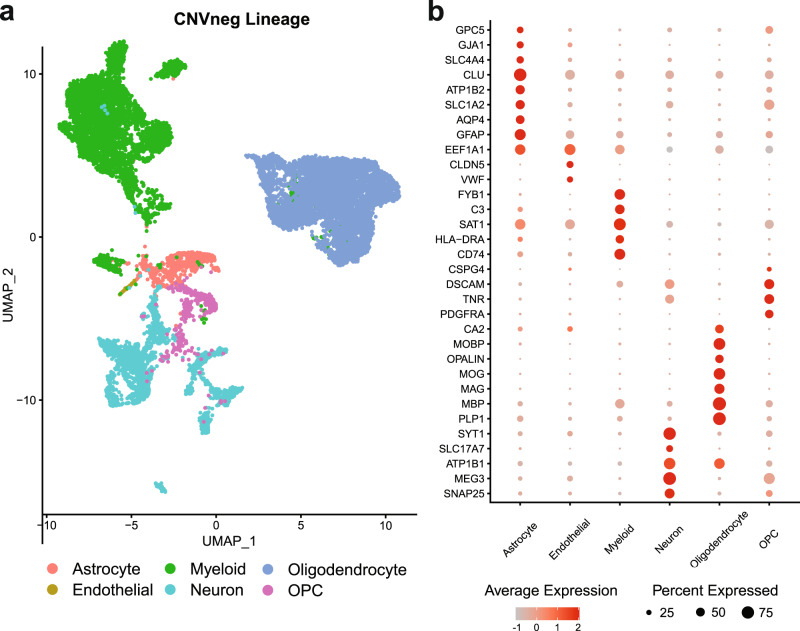
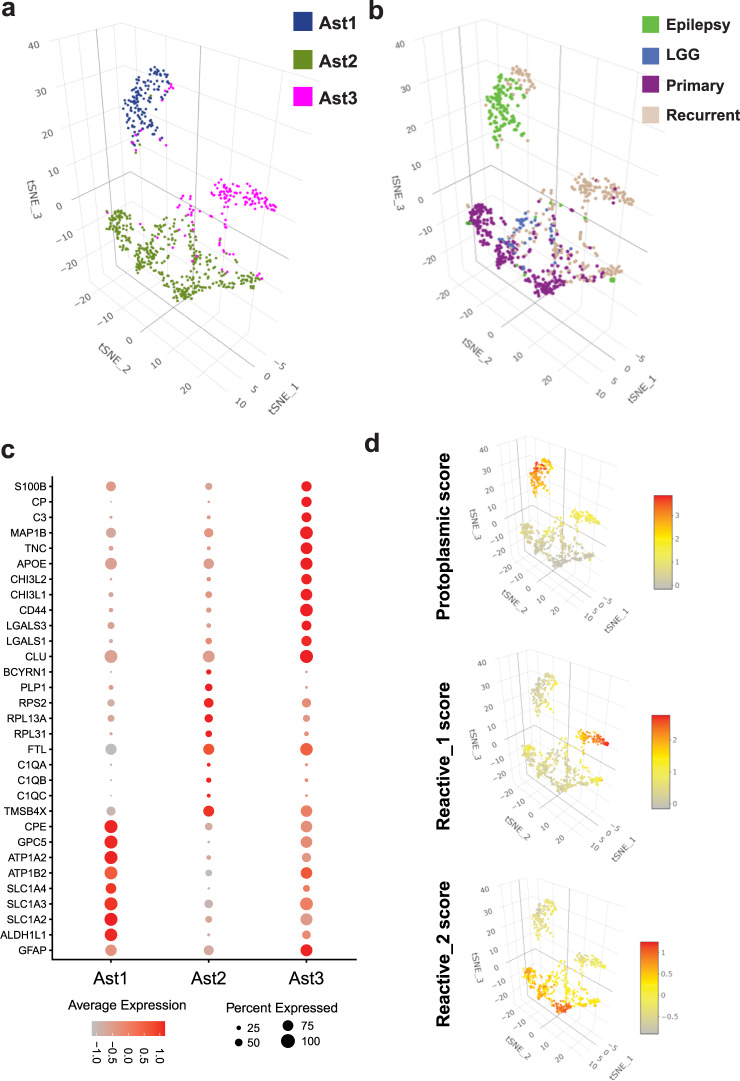
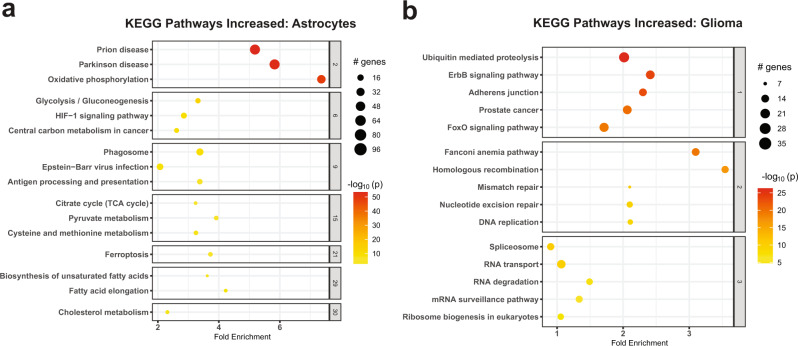
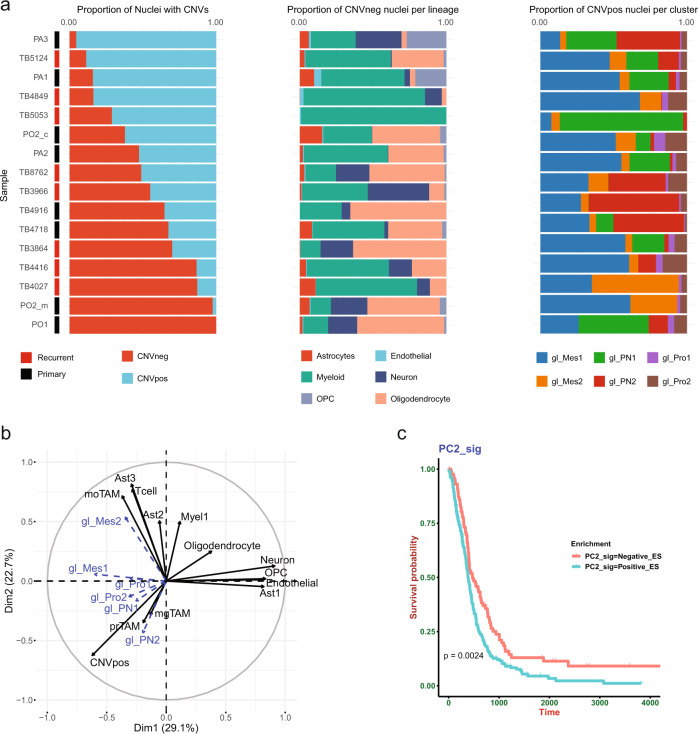
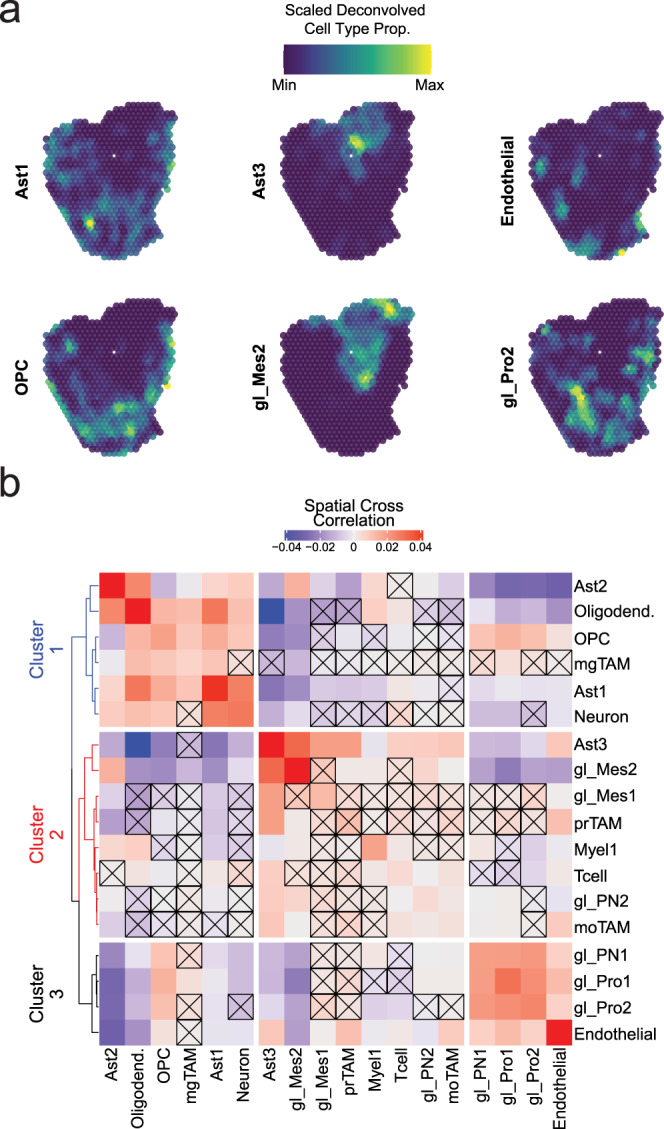
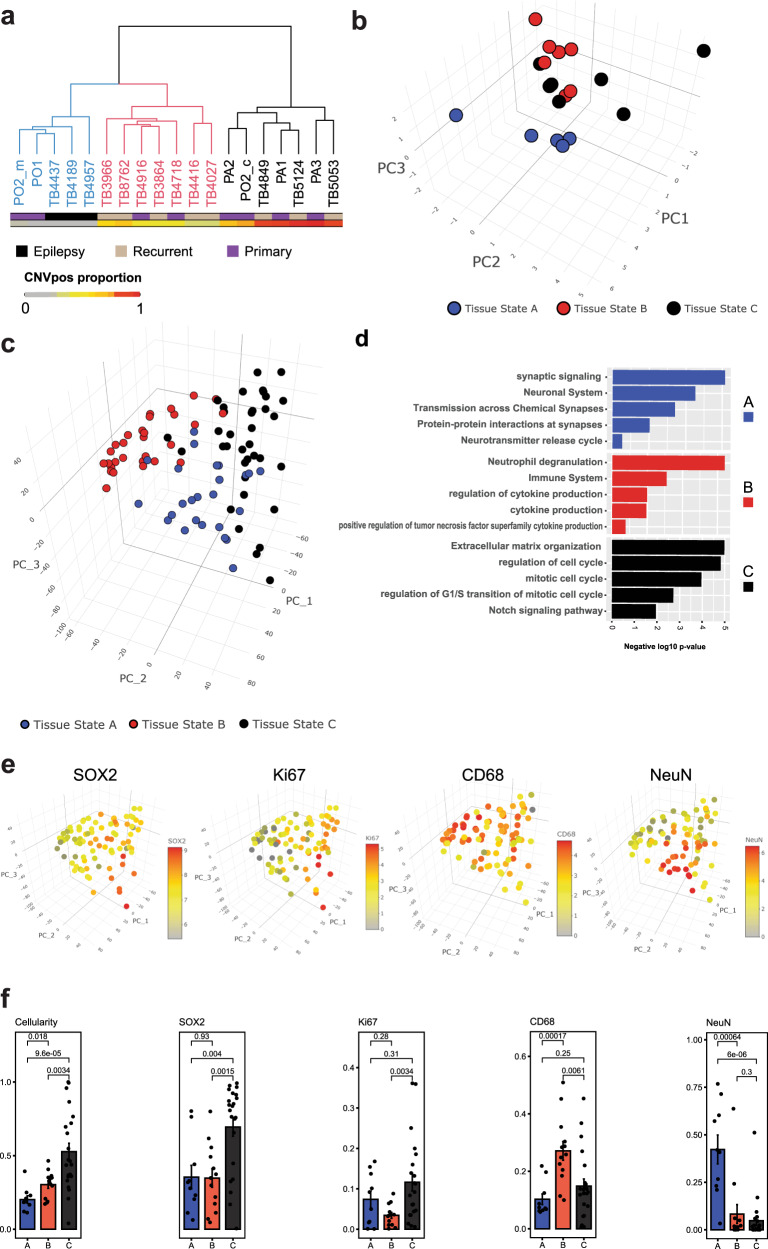
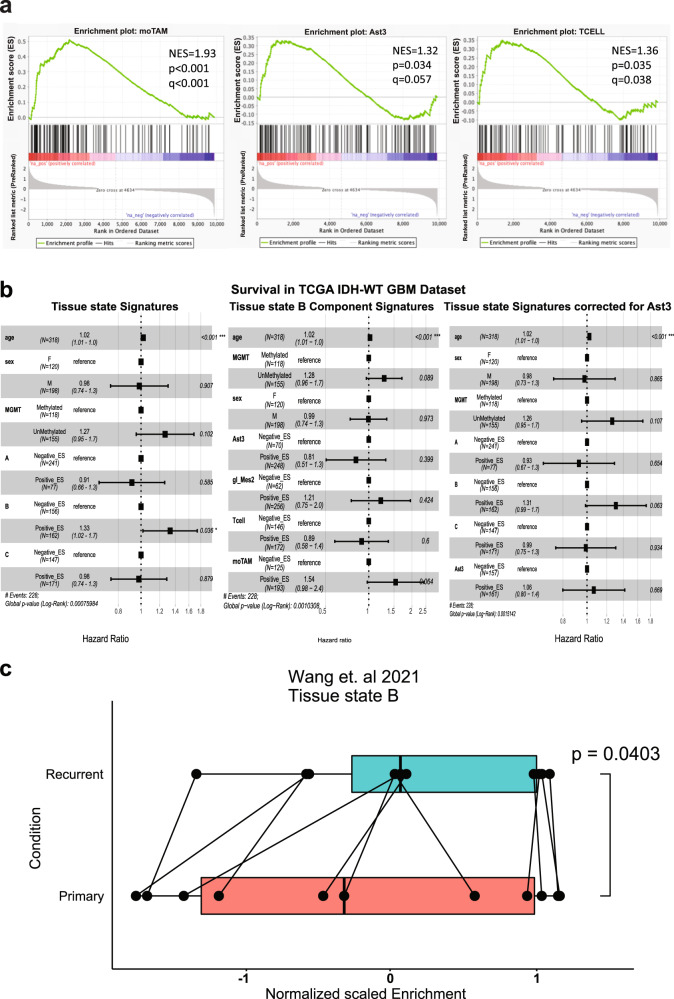
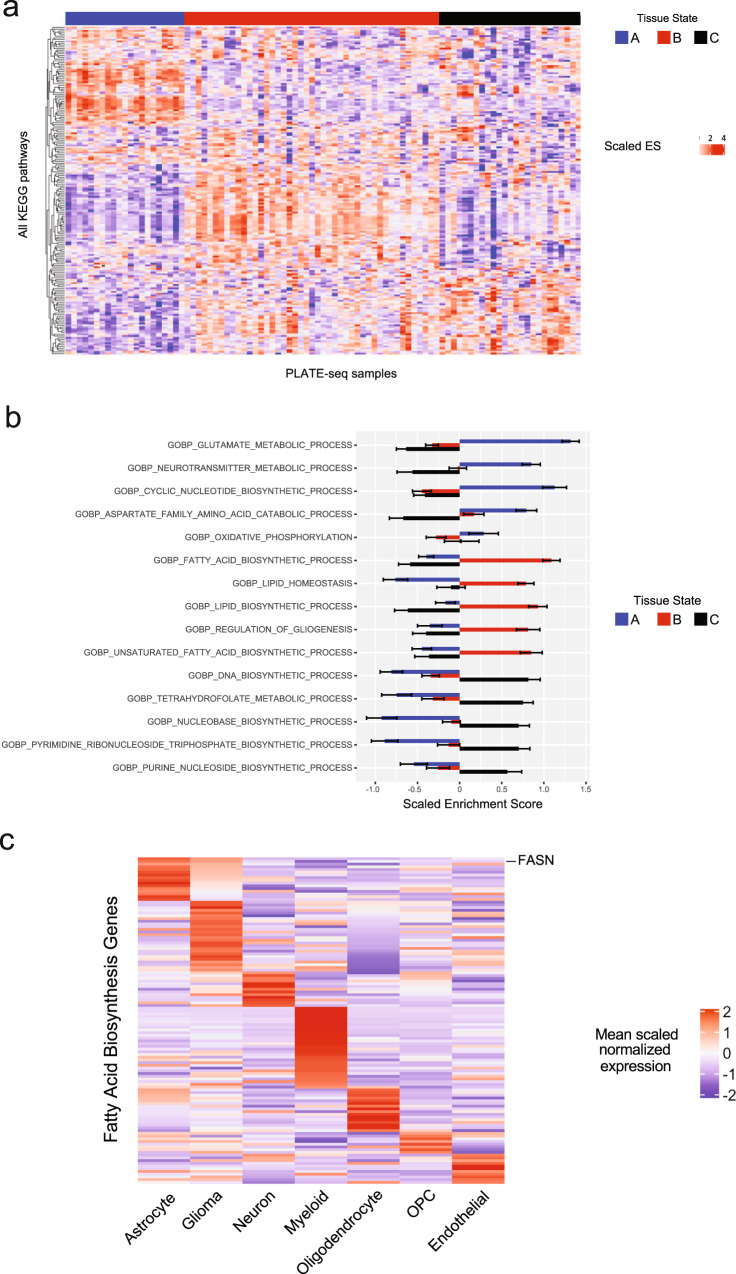
Glioblastoma (GBM) diffusely infiltrates the brain and intermingles with non-neoplastic brain cells, including astrocytes, neurons and microglia/myeloid cells. This complex mixture of cell types forms the biological context for therapeutic response and tumor recurrence. We used single-nucleus RNA sequencing and spatial transcriptomics to determine the cellular composition and transcriptional states in primary and recurrent glioma and identified three compositional 'tissue-states' defined by cohabitation patterns between specific subpopulations of neoplastic and non-neoplastic brain cells. These tissue-states correlated with radiographic, histopathologic, and prognostic features and were enriched in distinct metabolic pathways. Fatty acid biosynthesis was enriched in the tissue-state defined by the cohabitation of astrocyte-like/mesenchymal glioma cells, reactive astrocytes, and macrophages, and was associated with recurrent GBM and shorter survival. Treating acute slices of GBM with a fatty acid synthesis inhibitor depleted the transcriptional signature of this pernicious tissue-state. These findings point to therapies that target interdependencies in the GBM microenvironment.
© 2023. The Author(s).
Conflict of interest statement
P.A.S. receives patent royalties from Guardant Health. Columbia University has filed a patent application on the microwell single-cell RNA-seq technology used in this study, and P.A.S. is listed as a co-inventor. The patent number is WO/2016/191533. The patent title is “RNA printing and sequencing devices, methods, and systems”. None of the other authors declare any competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
