

Genomic mutation landscape of skin cancers from DNA repair-deficient xeroderma pigmentosum patients
- PMID: 37142601
- PMCID: PMC10160032
- DOI: 10.1038/s41467-023-38311-0
Genomic mutation landscape of skin cancers from DNA repair-deficient xeroderma pigmentosum patients
Abstract
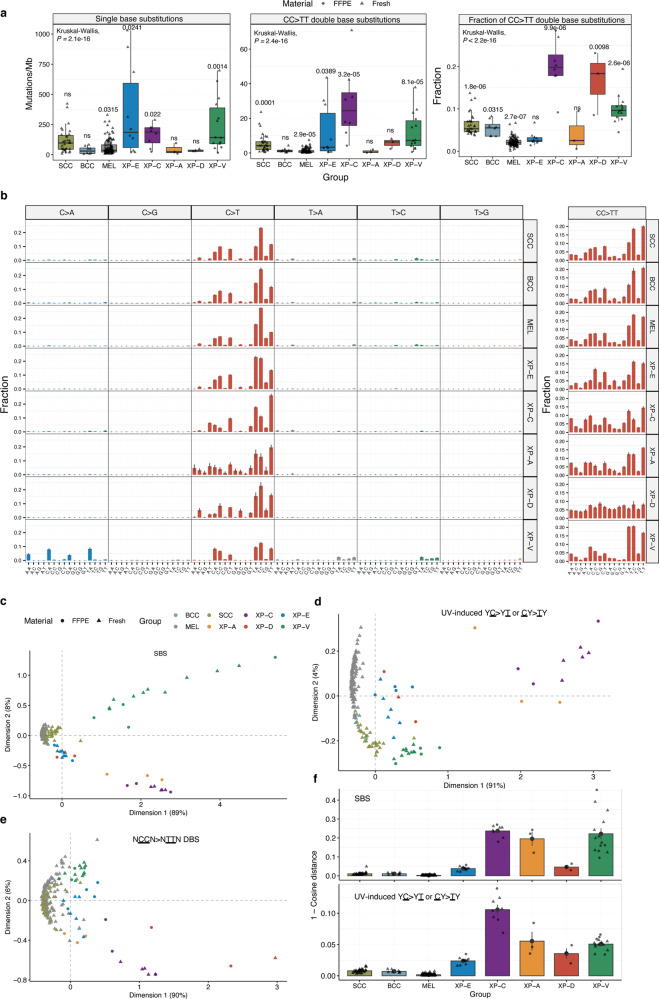
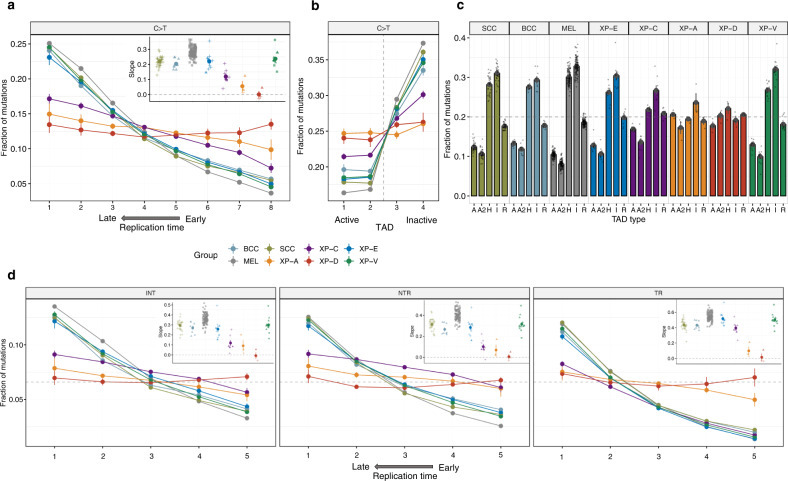
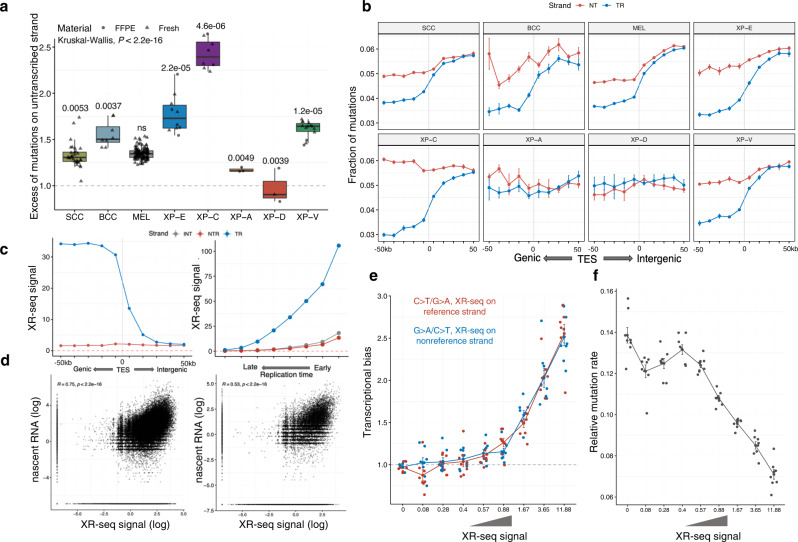
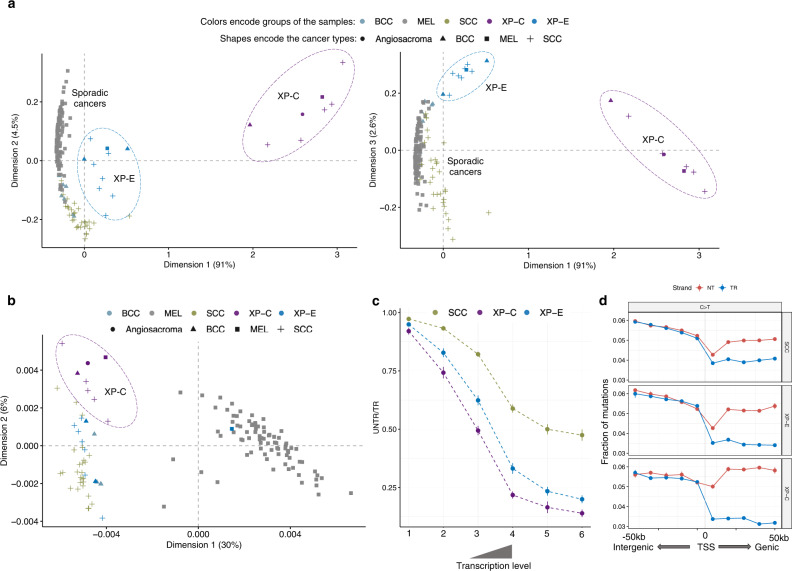
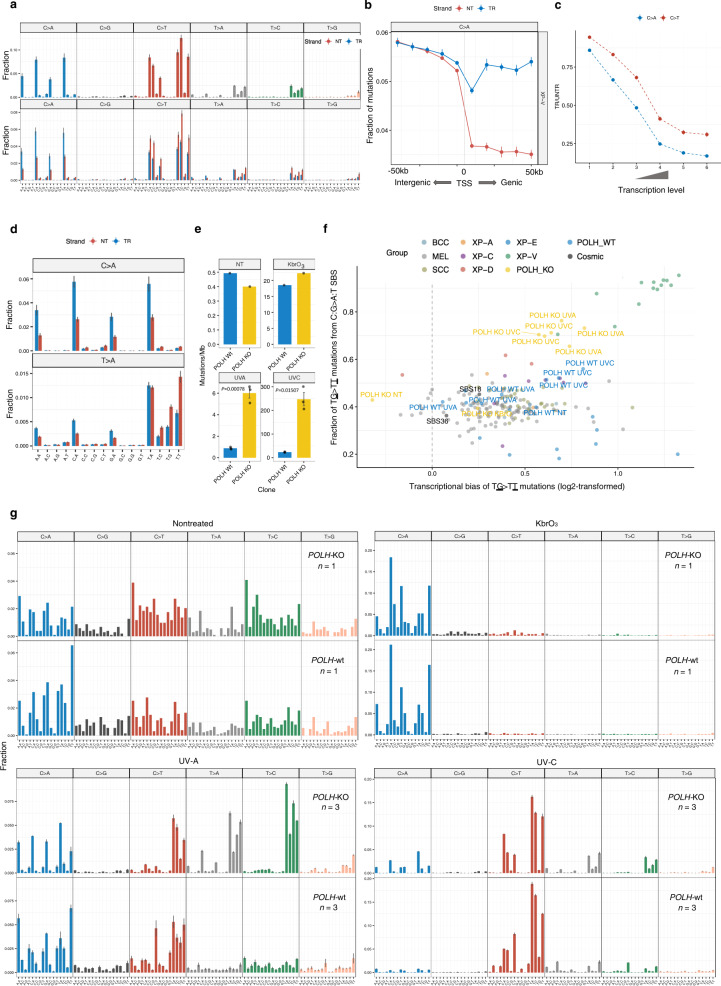
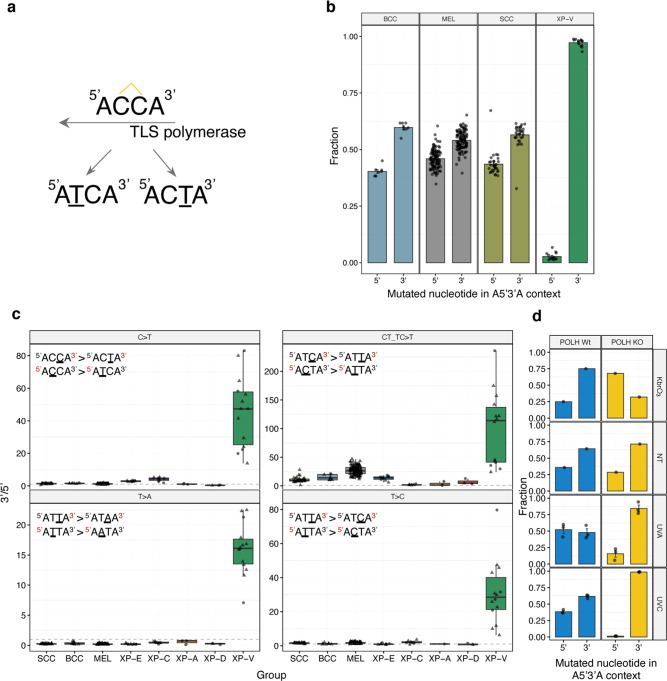
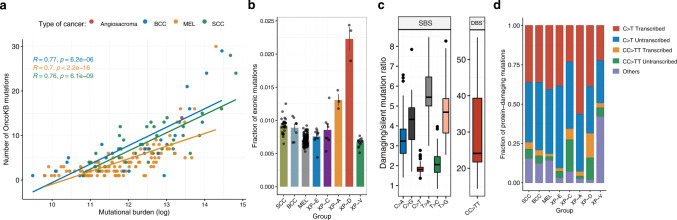
Xeroderma pigmentosum (XP) is a genetic disorder caused by mutations in genes of the Nucleotide Excision Repair (NER) pathway (groups A-G) or in Translesion Synthesis DNA polymerase η (V). XP is associated with an increased skin cancer risk, reaching, for some groups, several thousand-fold compared to the general population. Here, we analyze 38 skin cancer genomes from five XP groups. We find that the activity of NER determines heterogeneity of the mutation rates across skin cancer genomes and that transcription-coupled NER extends beyond the gene boundaries reducing the intergenic mutation rate. Mutational profile in XP-V tumors and experiments with POLH knockout cell line reveal the role of polymerase η in the error-free bypass of (i) rare TpG and TpA DNA lesions, (ii) 3' nucleotides in pyrimidine dimers, and (iii) TpT photodimers. Our study unravels the genetic basis of skin cancer risk in XP and provides insights into the mechanisms reducing UV-induced mutagenesis in the general population.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
