A human model of asthma exacerbation reveals transcriptional programs and cell circuits specific to allergic asthma
- PMID: 37146132
- PMCID: PMC10440046
- DOI: 10.1126/sciimmunol.abq6352
A human model of asthma exacerbation reveals transcriptional programs and cell circuits specific to allergic asthma
Abstract
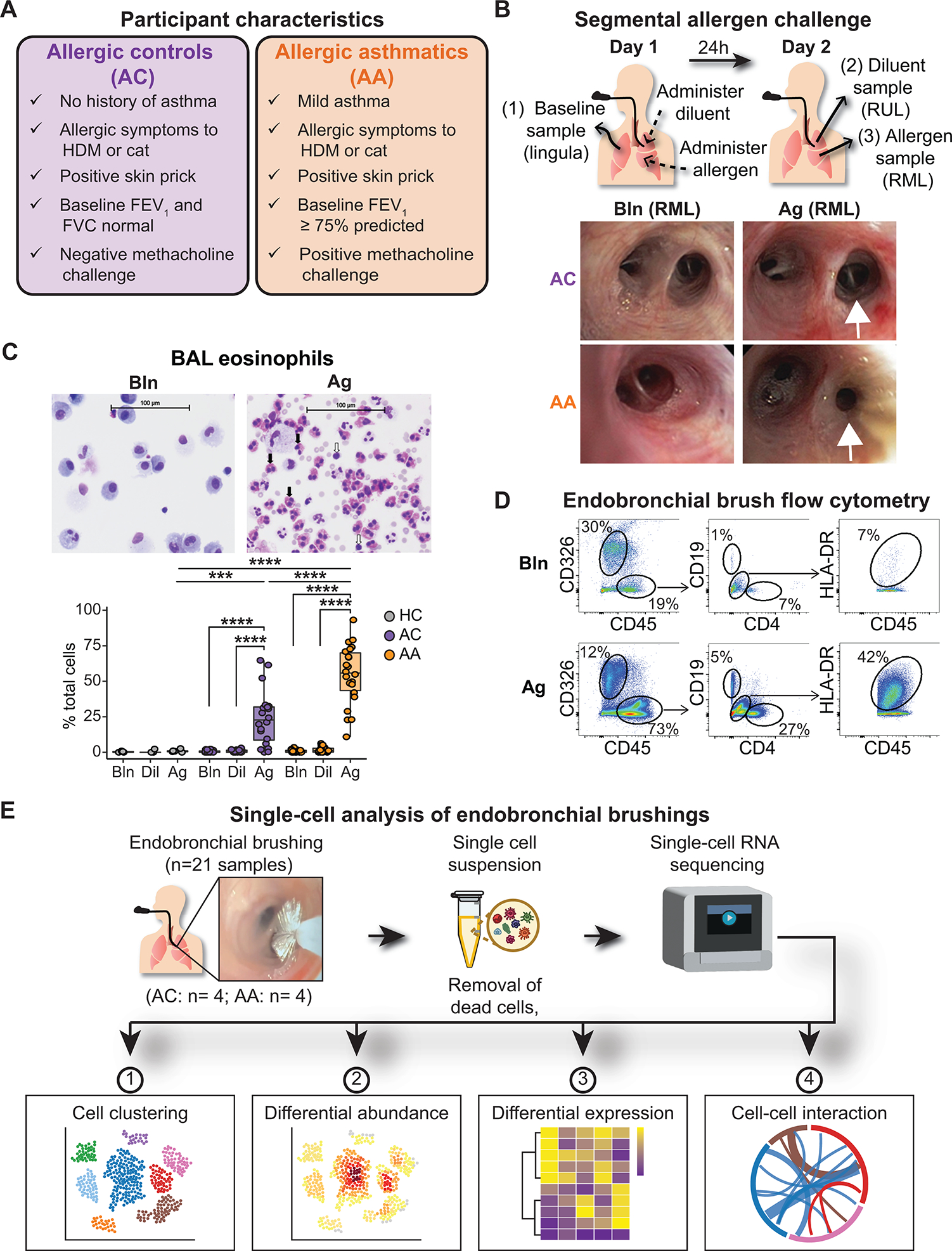
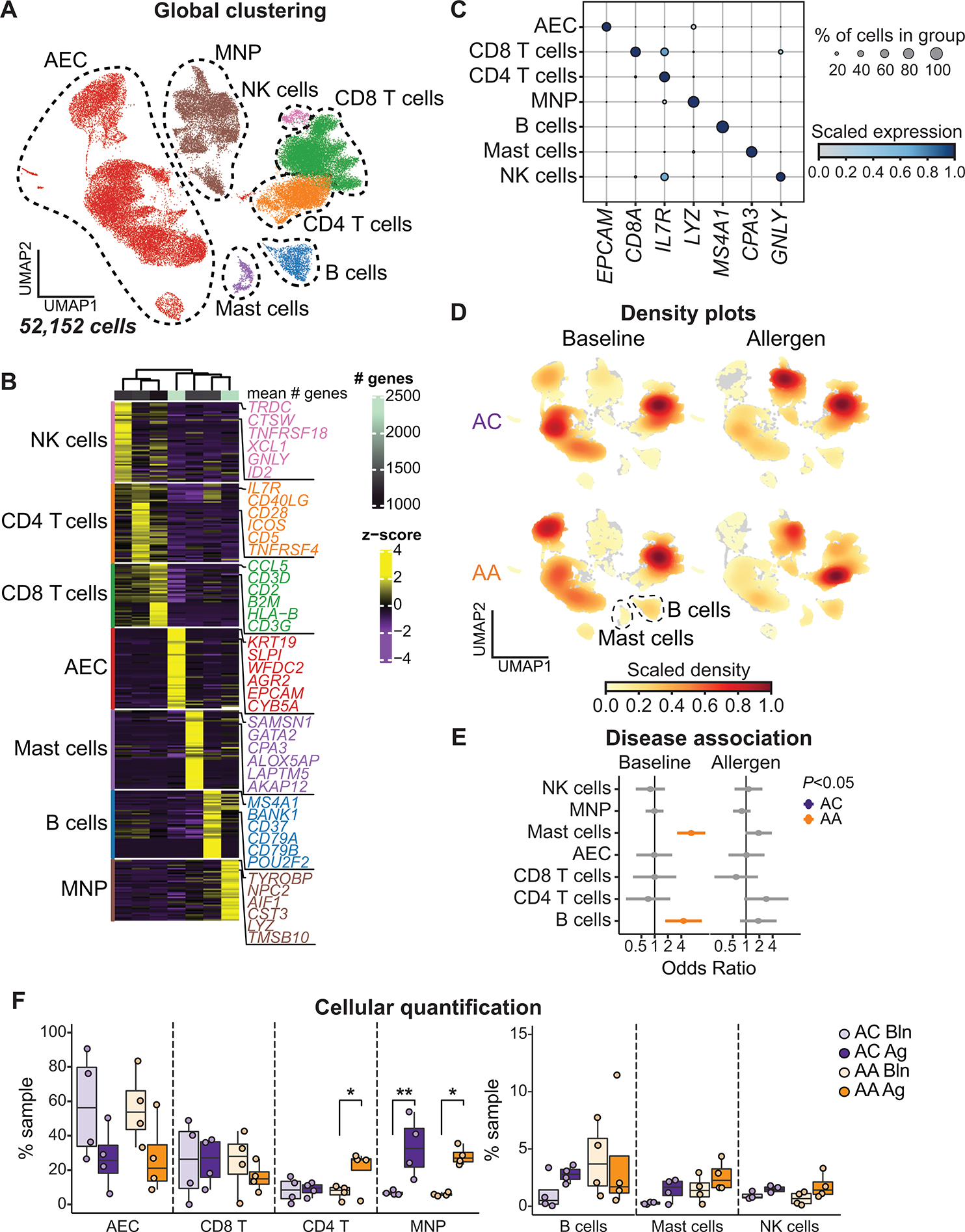
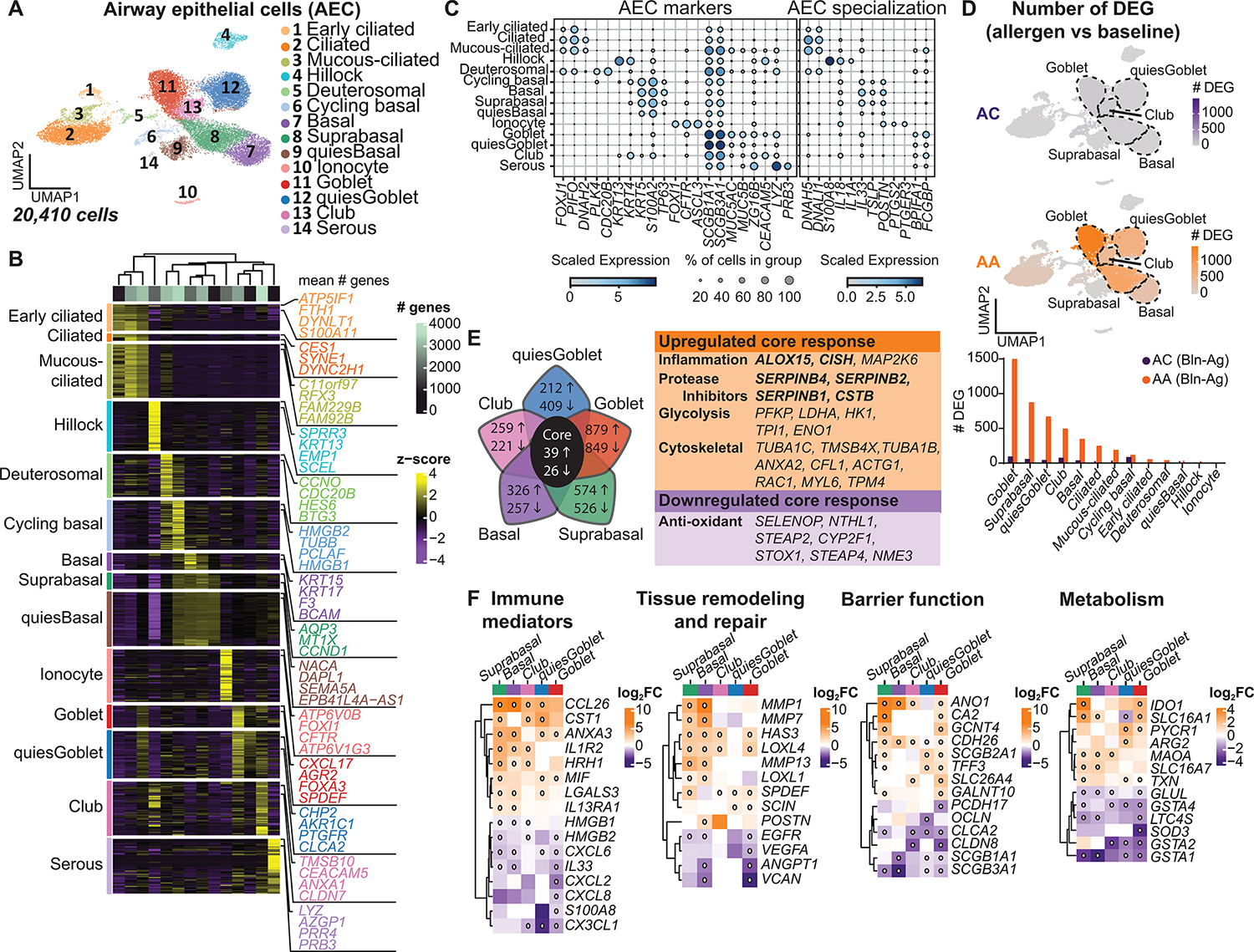
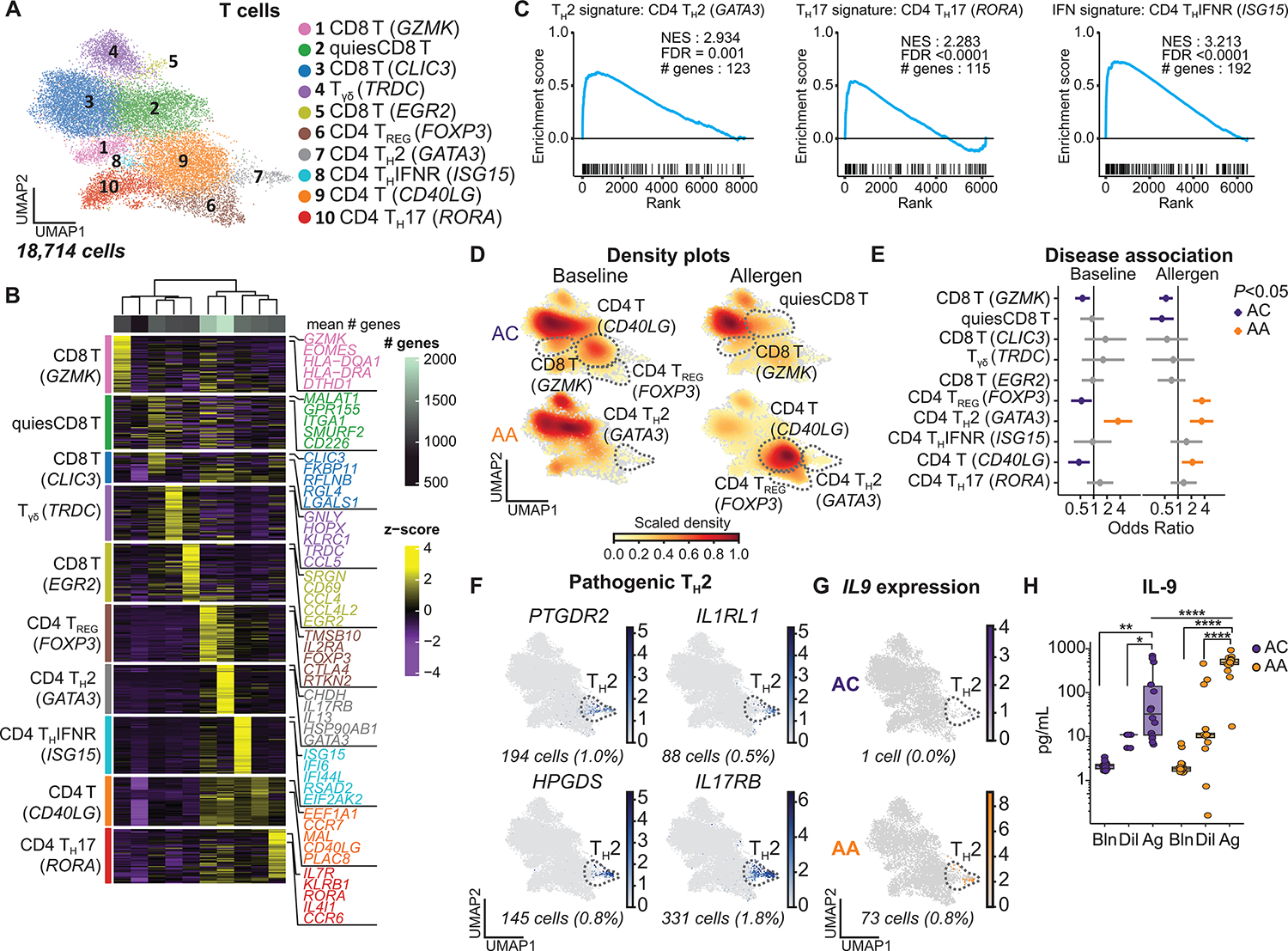
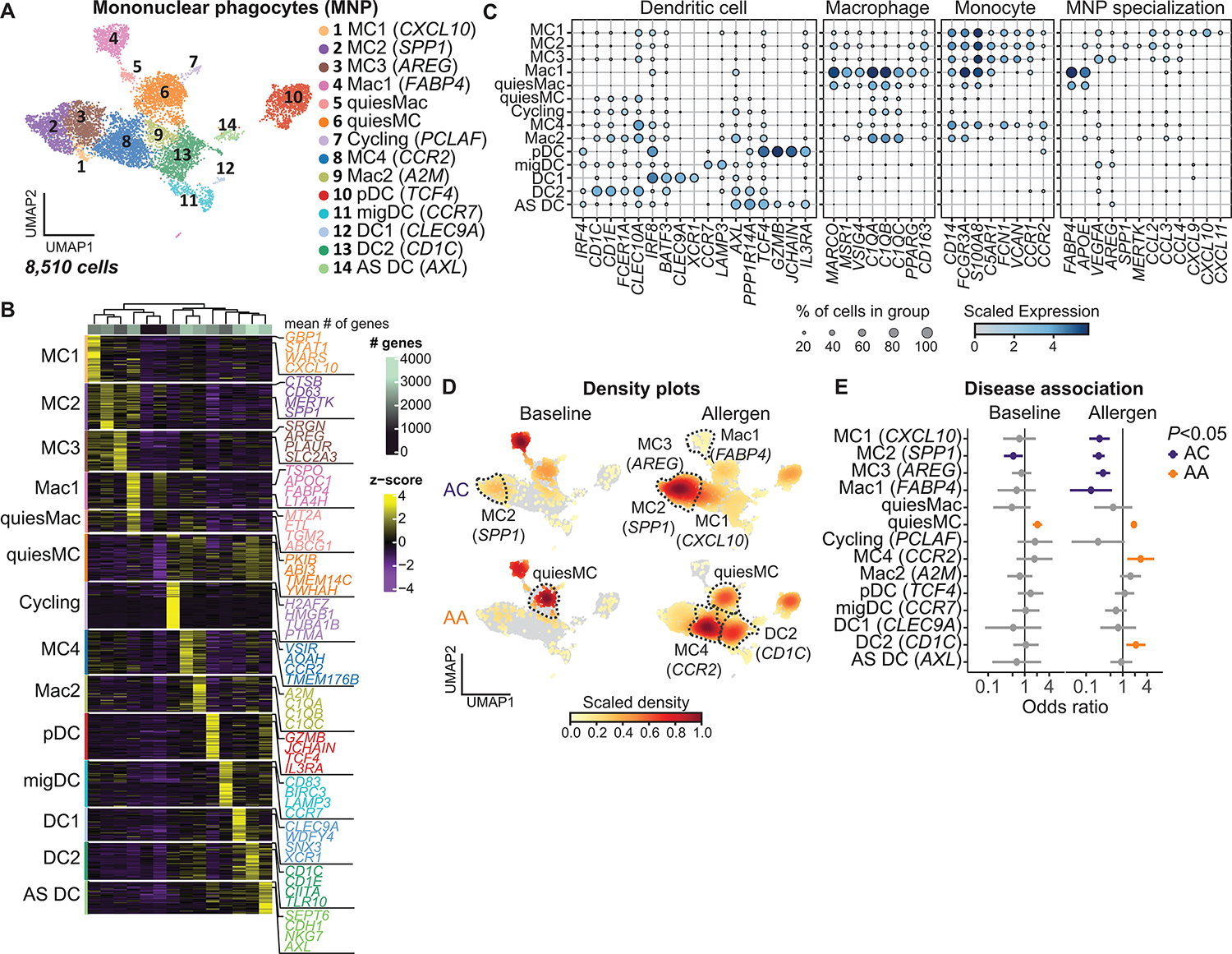
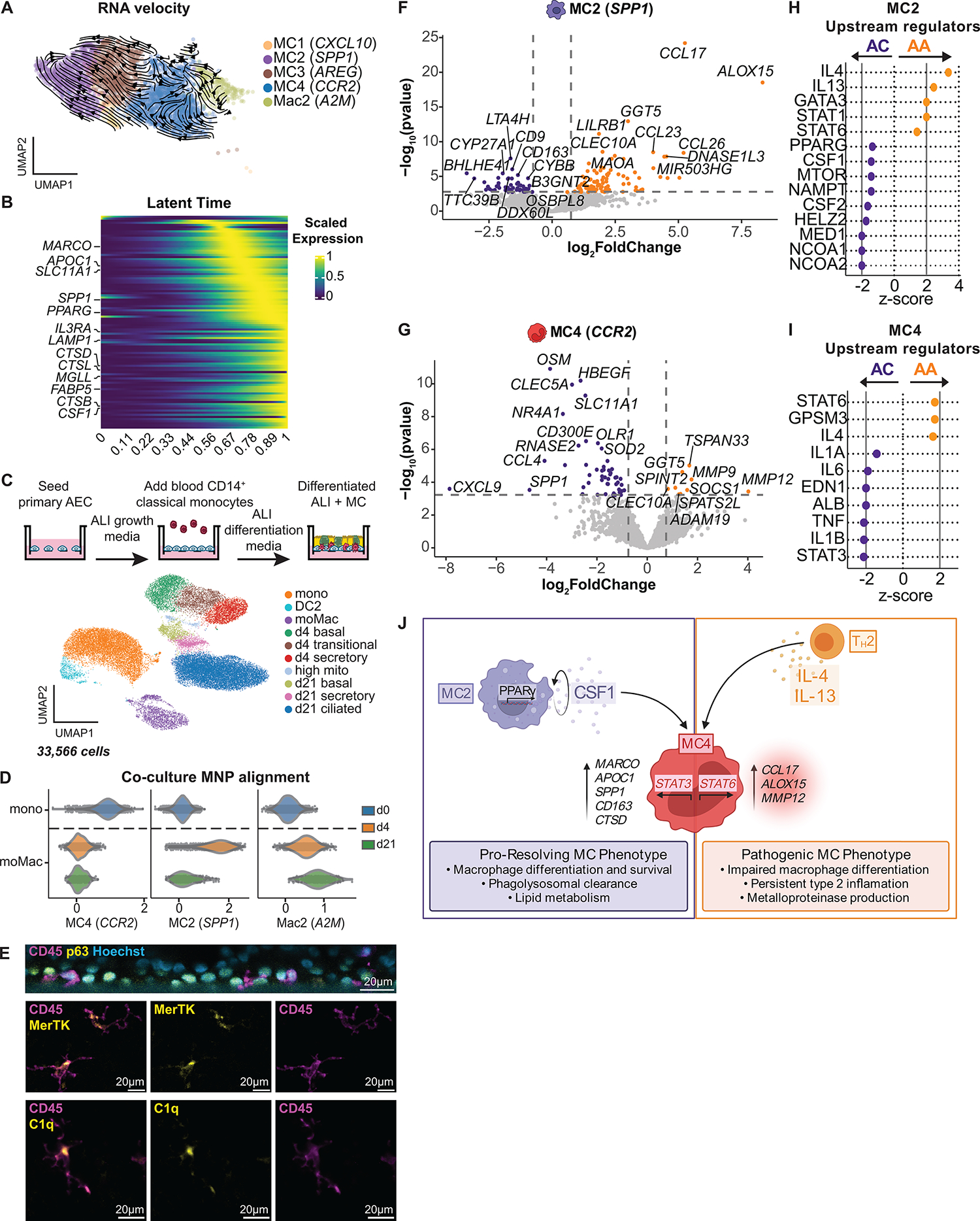
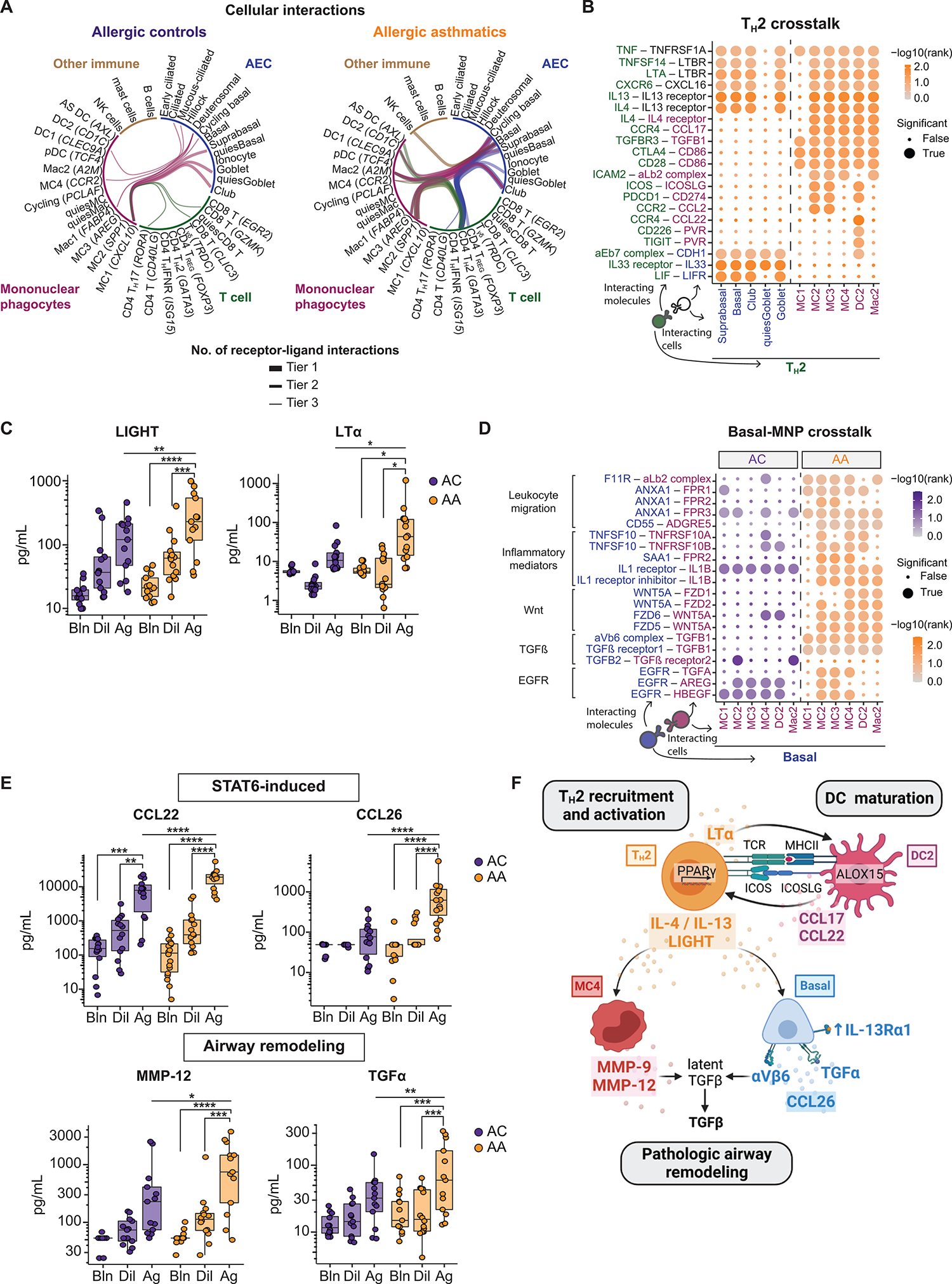
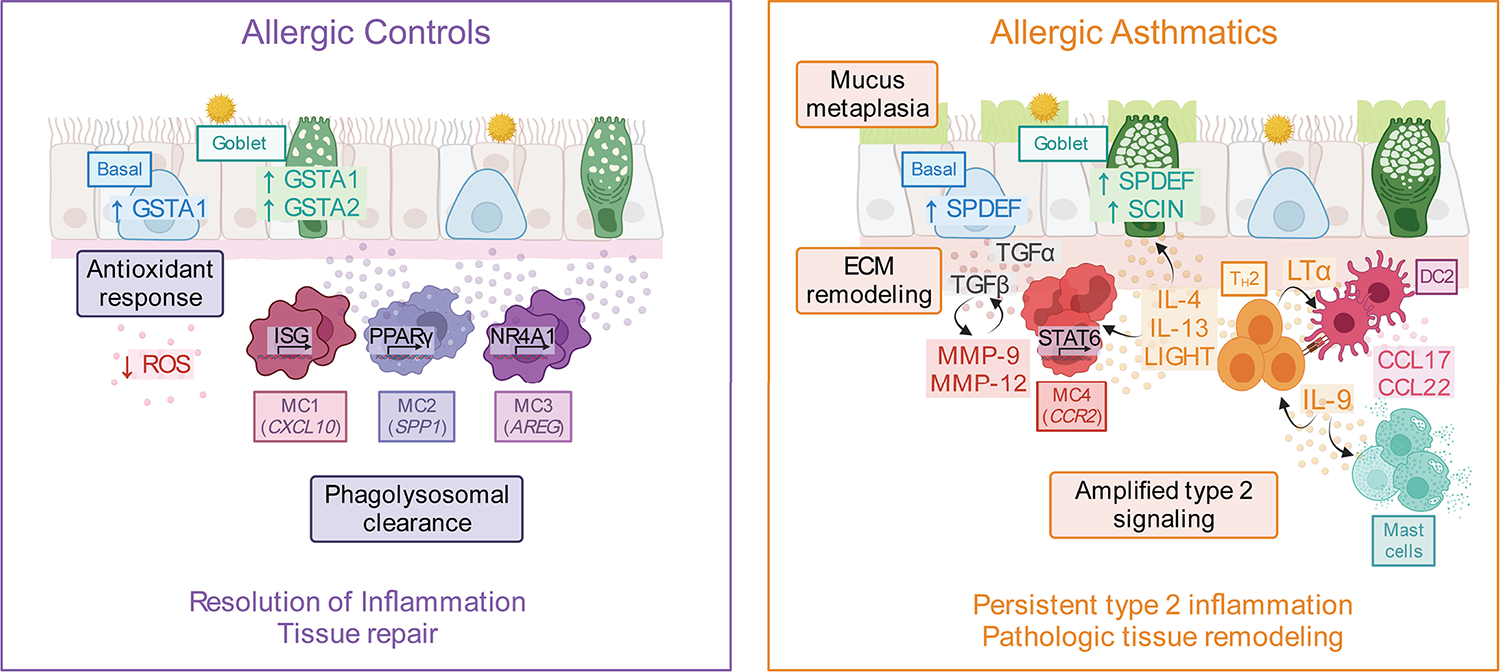
Asthma is a chronic disease most commonly associated with allergy and type 2 inflammation. However, the mechanisms that link airway inflammation to the structural changes that define asthma are incompletely understood. Using a human model of allergen-induced asthma exacerbation, we compared the lower airway mucosa in allergic asthmatics and allergic non-asthmatic controls using single-cell RNA sequencing. In response to allergen, the asthmatic airway epithelium was highly dynamic and up-regulated genes involved in matrix degradation, mucus metaplasia, and glycolysis while failing to induce injury-repair and antioxidant pathways observed in controls. IL9-expressing pathogenic TH2 cells were specific to asthmatic airways and were only observed after allergen challenge. Additionally, conventional type 2 dendritic cells (DC2 that express CD1C) and CCR2-expressing monocyte-derived cells (MCs) were uniquely enriched in asthmatics after allergen, with up-regulation of genes that sustain type 2 inflammation and promote pathologic airway remodeling. In contrast, allergic controls were enriched for macrophage-like MCs that up-regulated tissue repair programs after allergen challenge, suggesting that these populations may protect against asthmatic airway remodeling. Cellular interaction analyses revealed a TH2-mononuclear phagocyte-basal cell interactome unique to asthmatics. These pathogenic cellular circuits were characterized by type 2 programming of immune and structural cells and additional pathways that may sustain and amplify type 2 signals, including TNF family signaling, altered cellular metabolism, failure to engage antioxidant responses, and loss of growth factor signaling. Our findings therefore suggest that pathogenic effector circuits and the absence of proresolution programs drive structural airway disease in response to type 2 inflammation.
Conflict of interest statement
Figures
Comment in
-
See no allergen, hear no allergen, speak no allergen!Sci Immunol. 2023 May 12;8(83):eadh0597. doi: 10.1126/sciimmunol.adh0597. Epub 2023 May 5. Sci Immunol. 2023. PMID: 37146130
References
-
- Centers for Disease Control and Prevention, CDC Virtal Signs—Asthma in the US (Centers for Disease Control and Prevention, 2011); www.cdc.gov/vitalsigns/asthma/index.html.
-
- Hammad H, Lambrecht BN, The basic immunology of asthma. Cell 184, 1469–1485 (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AI168131/AI/NIAID NIH HHS/United States
- KL2 TR002542/TR/NCATS NIH HHS/United States
- UL1 TR002541/TR/NCATS NIH HHS/United States
- R21 AI173732/AI/NIAID NIH HHS/United States
- R01 AI040618/AI/NIAID NIH HHS/United States
- K08 HL140173/HL/NHLBI NIH HHS/United States
- UH2 AI144434/AI/NIAID NIH HHS/United States
- R03 HL164370/HL/NHLBI NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- P30 ES005605/ES/NIEHS NIH HHS/United States
- T32 AR007258/AR/NIAMS NIH HHS/United States
- DP2 CA247831/CA/NCI NIH HHS/United States
- P30 DK054759/DK/NIDDK NIH HHS/United States
- T32 HL116275/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases