

Case report: Neonatal autoimmune lymphoproliferative syndrome with a novel pathogenic homozygous FAS variant effectively treated with sirolimus
- PMID: 37152306
- PMCID: PMC10159173
- DOI: 10.3389/fped.2023.1150179
Case report: Neonatal autoimmune lymphoproliferative syndrome with a novel pathogenic homozygous FAS variant effectively treated with sirolimus
Abstract
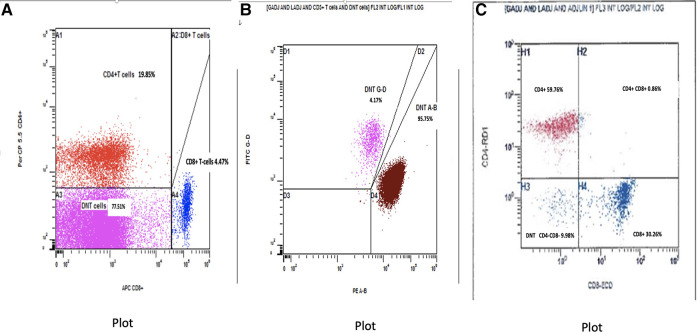
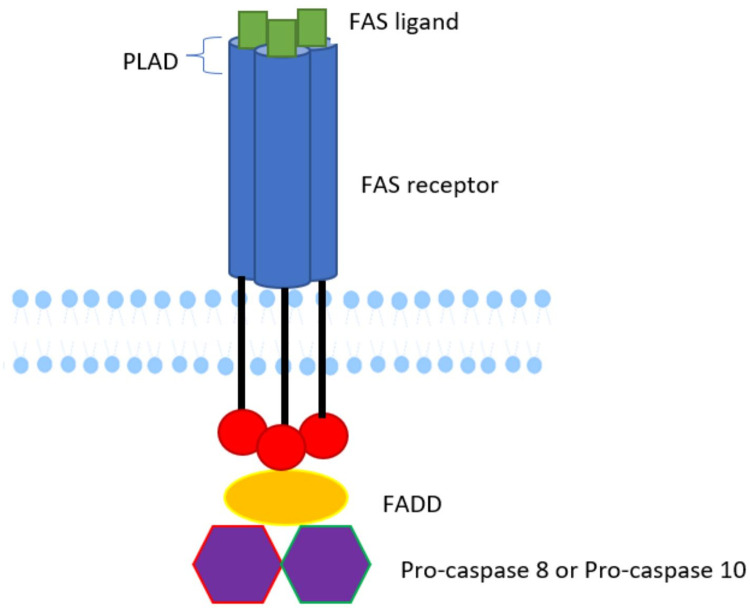
Background: Autoimmune lymphoproliferative syndrome (ALPS) is a rare disease characterized by defective FAS signaling, which results in chronic, nonmalignant lymphoproliferation and autoimmunity accompanied by increased numbers of "double-negative" T-cells (DNTs) (T-cell receptor αβ+ CD4-CD8-) and an increased risk of developing malignancies later in life.
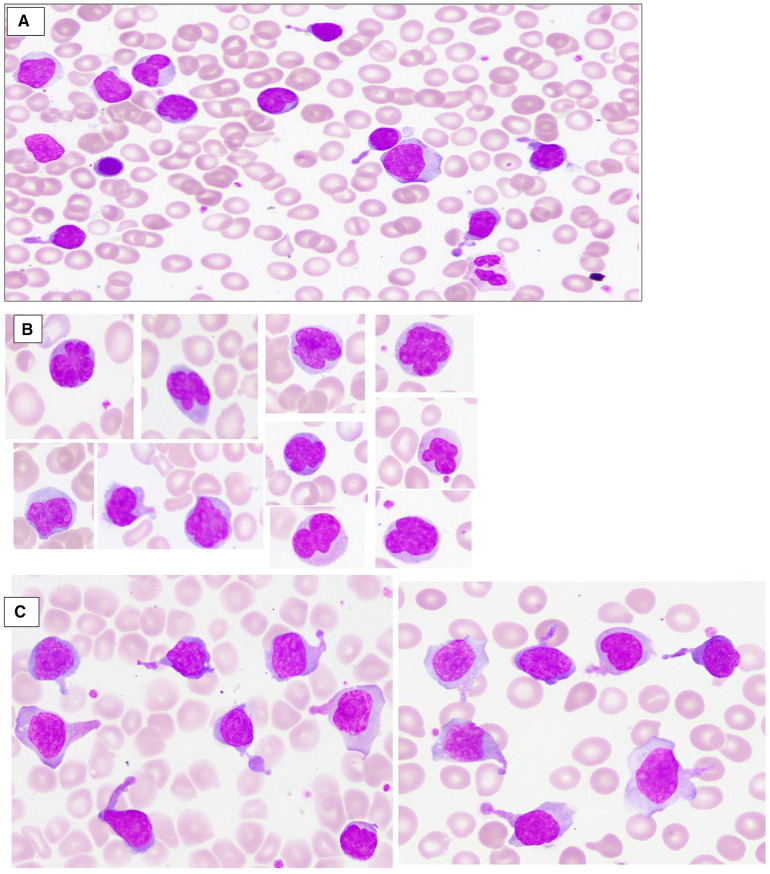
Case presentation: We herein report a case of a newborn boy with a novel germline homozygous variant identified in the FAS gene, exon 9, c.775del, which was considered pathogenic. The consequence of this sequence change was the creation of a premature translational stop signal p.(lle259*), associated with a severe clinical phenotype of ALPS-FAS. The elder brother of the proband was also affected by ALPS and has been found to have the same FAS homozygous variant associated with a severe clinical phenotype of ALPS-FAS, whereas the unaffected parents are heterozygous carriers of this variant. This new variant has not previously been described in population databases (gnomAD and ExAC) or in patients with FAS-related conditions. Treatment with sirolimus effectively improved the patient clinical manifestations with obvious reduction in the percentage of DNTs.
Conclusion: We described a new ALPS-FAS clinical phenotype-associated germline FAS homozygous pathogenic variant, exon 9, c.775del, that produces a premature translational stop signal p.(lle259*). Sirolimus significantly reduced DNTs and substantially relieved the patient's clinical symptoms.
Keywords: ALPS (autoimmune lymphoproliferative syndrome); DNT-cells; FAS; autosomal recessive; newborn; novel variant; sirolimus.
© 2023 Elgharbawy, Karim, Soliman, Hassan, Sudarsanan and Gad.
Conflict of interest statement
FME, DSS, AS, and AG are employed by Hamad Medical Corporation. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous