

doi: 10.1038/s41587-023-01773-0.
Epub 2023 May 8.
Fast and accurate protein structure search with Foldseek
Affiliations
- PMID: 37156916
- PMCID: PMC10869269
- DOI: 10.1038/s41587-023-01773-0
Item in Clipboard
Fast and accurate protein structure search with Foldseek
Nat Biotechnol.
2024 Feb.
Abstract
As structure prediction methods are generating millions of publicly available protein structures, searching these databases is becoming a bottleneck. Foldseek aligns the structure of a query protein against a database by describing tertiary amino acid interactions within proteins as sequences over a structural alphabet. Foldseek decreases computation times by four to five orders of magnitude with 86%, 88% and 133% of the sensitivities of Dali, TM-align and CE, respectively.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
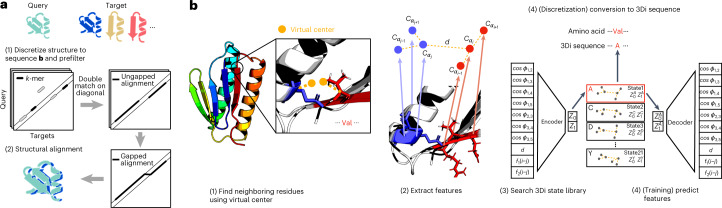
a, Foldseek searches a set of query structures through a set of target structures. (1) Query and target structures are discretized into 3Di sequences (see b). To detect candidate structures, we apply the fast and sensitive k-mer and ungapped alignment prefilter of MMseqs2 to the 3Di sequences, (2) followed by vectorized Smith–Waterman local alignment combining 3Di and amino acid substitution scores. Alternatively, a global alignment is computed with a 1.7-times accelerated TM-align version (Supplementary Fig. 12). b, Learning the 3Di alphabet. (1) 3Di states describe tertiary interaction between a residue i and its nearest neighbor j. Nearest neighbors have the closest virtual center distance (yellow). Virtual center positions (Supplementary Fig. 1) were optimized for maximum search sensitivity. (2) To describe the interaction geometry of residues i and j, we extract seven angles, the Euclidean Cα distance and two sequence distance features from the six Cα coordinates of the two backbone fragments (blue and red). (3) These 10 features are used to define 20 3Di states by training a VQ-VAE modified to learn states that are maximally evolutionary conserved. For structure searches, the encoder predicts the best-matching 3Di state for each residue.
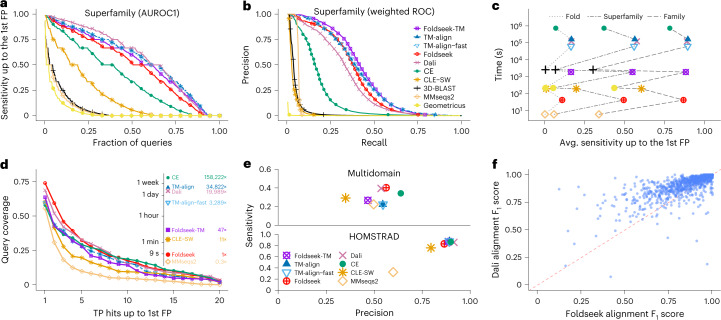
a, Cumulative distributions of sensitivity for homology detection on the SCOPe40 database of single-domain structures. TPs are matches within the same superfamily; FPs are matches between different folds. Sensitivity is the area under the ROC (AUROC) curve up to the first FP (see Supplementary Fig. 4 for family and fold). b, Precision-recall curve of SCOPe40 superfamilies (see Supplementary Fig. 4 for family and fold). c, Average sensitivity up to the first FP for family, superfamily and fold versus total runtime on an AMD EPYC 7702P 64-core CPU for the all-versus-all searches of 11,211 structures of SCOPe40. d, Search sensitivity on multi-domain, full-length AlphaFold2 protein models. One hundred queries, randomly selected from AlphaFoldDB (version 1), were searched against this database. Per-residue query coverage (y axis) is the fraction of residues covered by at least x (x axis) TP matches ranked before the first FP match. e, Alignment quality for alignments of AlphaFoldDB (version 1) protein models (top panel), averaged over the top five matches of each of the 100 queries. Sensitivity = TP residues in alignment / query length; precision = TP residues / alignment length. Reference-based alignment quality benchmark on HOMSTRAD alignments. f, Alignment quality comparison between Foldseek and Dali for each HOMSTRAD family. The F1 score is the harmonic mean between sensitivity and precision.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
