

TCR sequencing and cloning methods for repertoire analysis and isolation of tumor-reactive TCRs
- PMID: 37159666
- PMCID: PMC10163020
- DOI: 10.1016/j.crmeth.2023.100459
TCR sequencing and cloning methods for repertoire analysis and isolation of tumor-reactive TCRs
Abstract
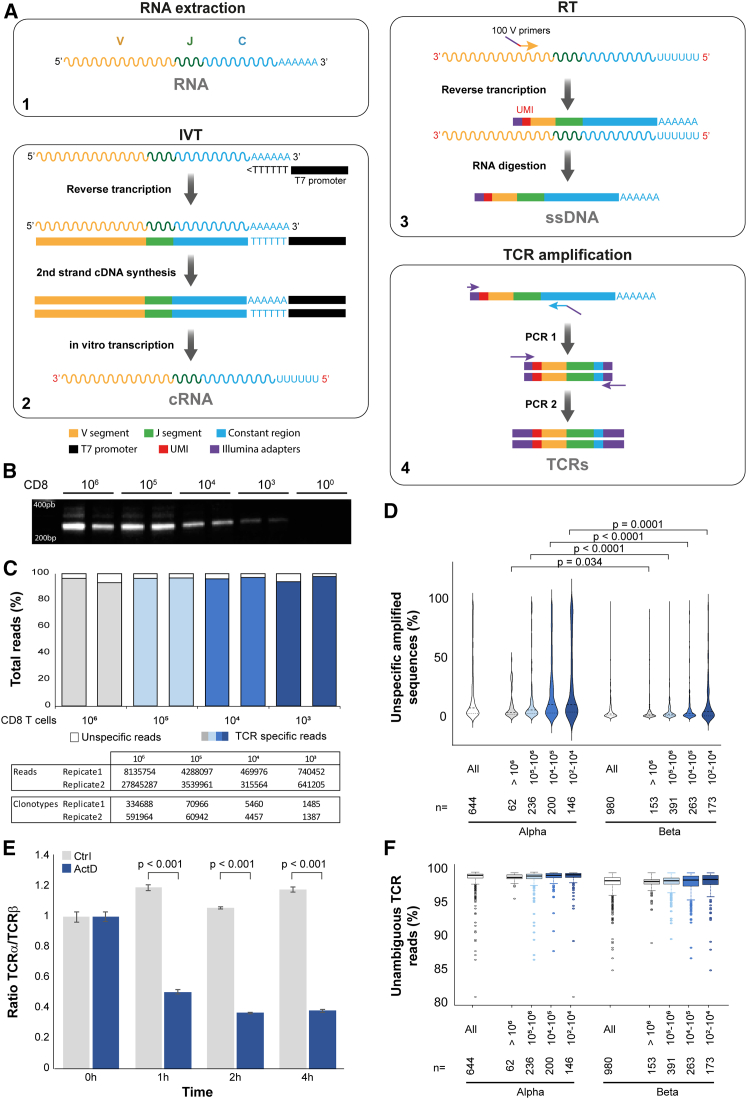
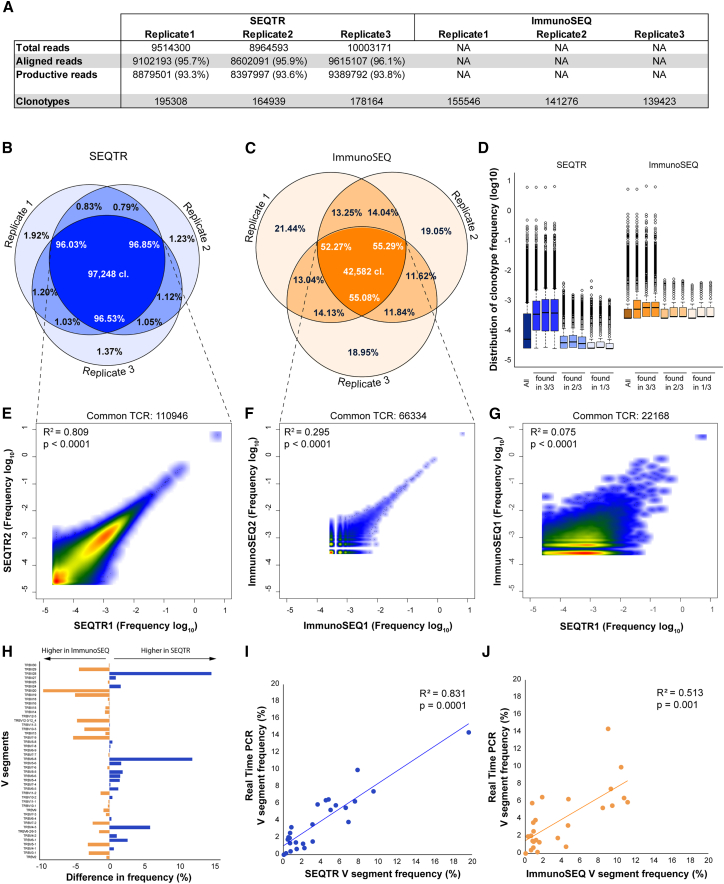
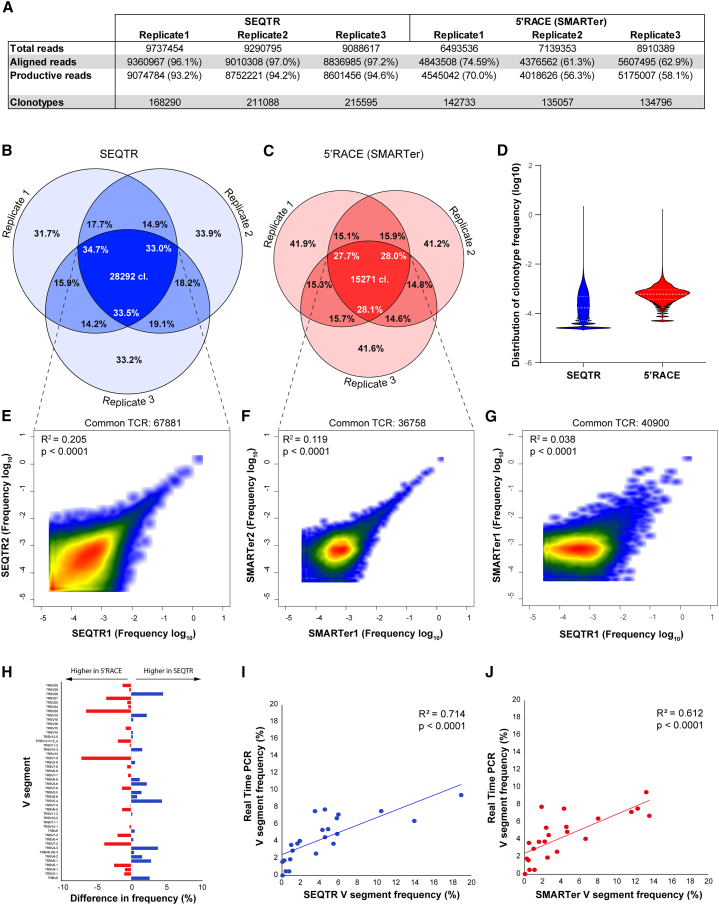
T cell receptor (TCR) technologies, including repertoire analyses and T cell engineering, are increasingly important in the clinical management of cellular immunity in cancer, transplantation, and other immune diseases. However, sensitive and reliable methods for repertoire analyses and TCR cloning are still lacking. Here, we report on SEQTR, a high-throughput approach to analyze human and mouse repertoires that is more sensitive, reproducible, and accurate as compared with commonly used assays, and thus more reliably captures the complexity of blood and tumor TCR repertoires. We also present a TCR cloning strategy to specifically amplify TCRs from T cell populations. Positioned downstream of single-cell or bulk TCR sequencing, it allows time- and cost-effective discovery, cloning, screening, and engineering of tumor-specific TCRs. Together, these methods will accelerate TCR repertoire analyses in discovery, translational, and clinical settings and permit fast TCR engineering for cellular therapies.
Keywords: T cell engineering; T cell receptor; TCR cloning; TCR repertoire; cancer; immune diversity; immune repertoire; immunotherapy; repertoire profiling.
© 2023 The Authors.
Conflict of interest statement
The University of Lausanne and Ludwig Institute for Cancer Research have filed for patent protection on the technology described herein. R.G. is named as inventor on this patent. G.C. has received grants from Celgene, Boehringer-Ingelheim, Roche, Bristol Myers Squibb, Iovance Therapeutics, and Kite Pharma. The institution with which G.C. is affiliated has received fees for G.C.’s participation on advisory boards or for presentation at a company-sponsored symposium from Genentech, Roche, Bristol Myers Squibb, AstraZeneca, NextCure, Geneos Tx, and Sanofi/Avensis. The Center Hospitalier Universitaire Vaudois (CHUV) and the Ludwig Institute for Cancer Research have filed for patent protection on the technology related to T cell expansion. S.B., A.H., and G.C. are named as inventors on this patent. G.C. has patents in the domain of antibodies and vaccines targeting the tumor vasculature as well as technologies related to T cell engineering for T cell therapy. G.C. holds patents around antibodies and receives royalties from the University of Pennsylvania regarding technology licensed to Novartis.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
