

Comparison of tumor-informed and tumor-naïve sequencing assays for ctDNA detection in breast cancer
- PMID: 37161793
- PMCID: PMC10245040
- DOI: 10.15252/emmm.202216505
Comparison of tumor-informed and tumor-naïve sequencing assays for ctDNA detection in breast cancer
Abstract
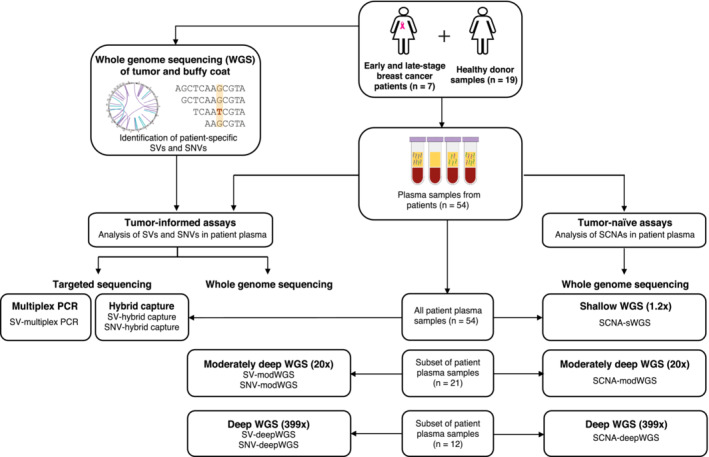
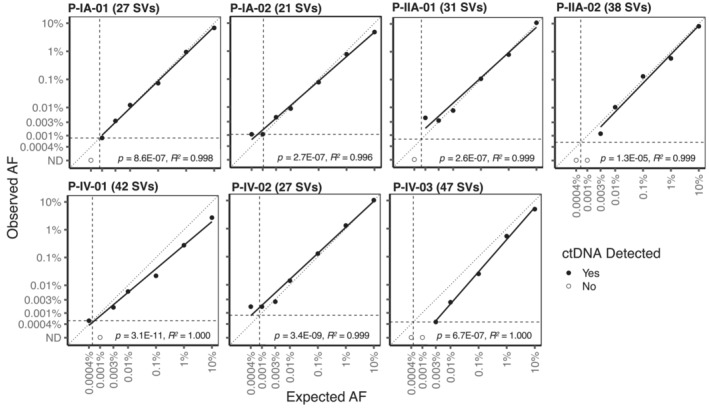
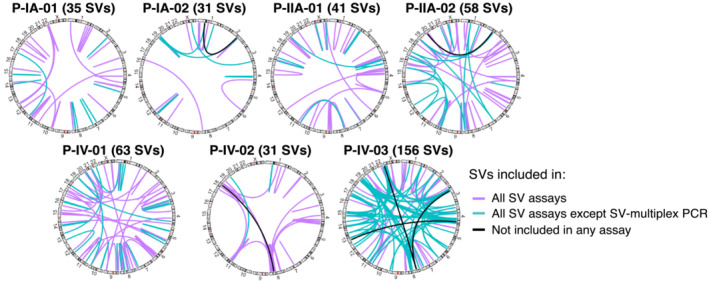
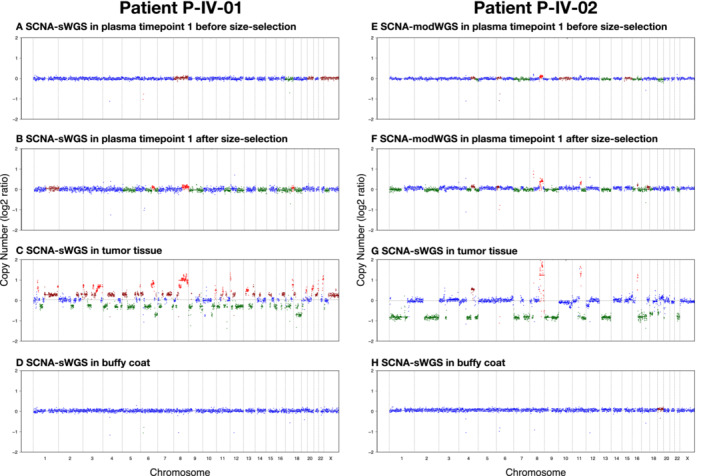
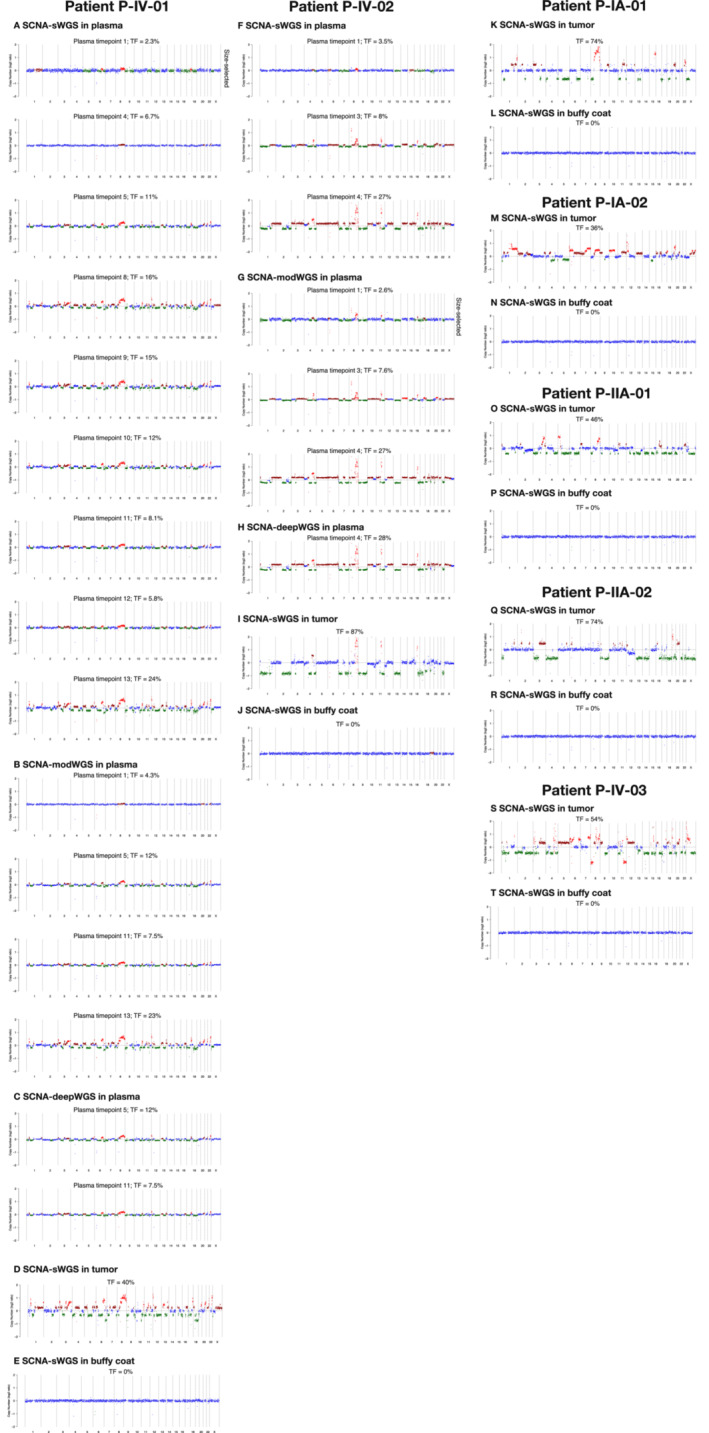
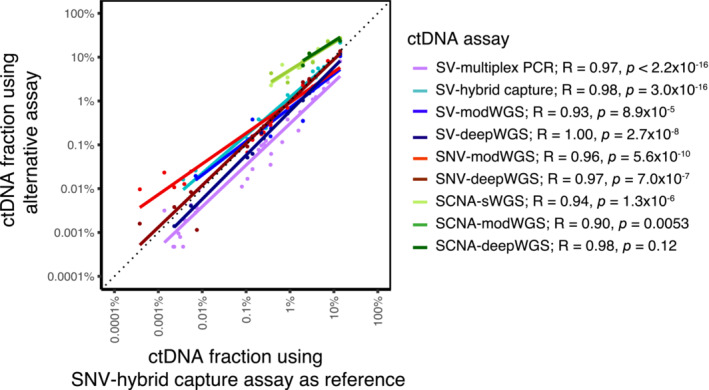
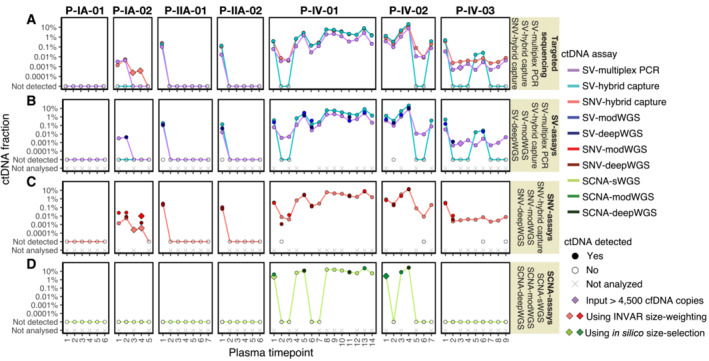
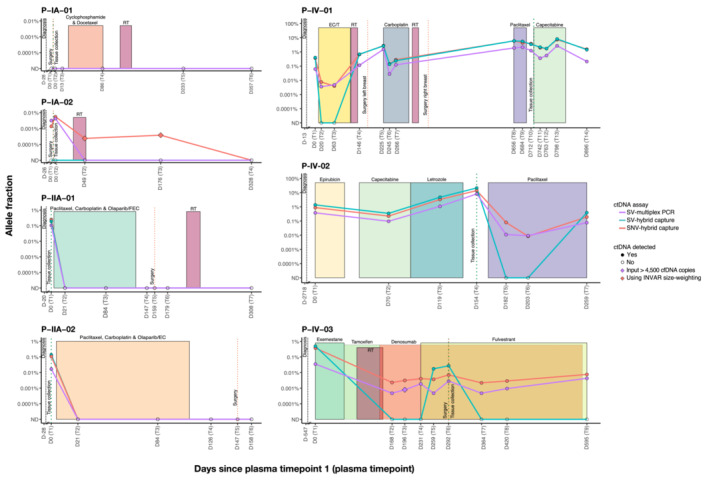
Analysis of circulating tumor DNA (ctDNA) to monitor cancer dynamics and detect minimal residual disease has been an area of increasing interest. Multiple methods have been proposed but few studies have compared the performance of different approaches. Here, we compare detection of ctDNA in serial plasma samples from patients with breast cancer using different tumor-informed and tumor-naïve assays designed to detect structural variants (SVs), single nucleotide variants (SNVs), and/or somatic copy-number aberrations, by multiplex PCR, hybrid capture, and different depths of whole-genome sequencing. Our results demonstrate that the ctDNA dynamics and allele fractions (AFs) were highly concordant when analyzing the same patient samples using different assays. Tumor-informed assays showed the highest sensitivity for detection of ctDNA at low concentrations. Hybrid capture sequencing targeting between 1,347 and 7,491 tumor-identified mutations at high depth was the most sensitive assay, detecting ctDNA down to an AF of 0.00024% (2.4 parts per million, ppm). Multiplex PCR targeting 21-47 tumor-identified SVs per patient detected ctDNA down to 0.00047% AF (4.7 ppm) and has potential as a clinical assay.
Keywords: circulating tumor DNA; hybrid capture; liquid biopsy; multiplex PCR; whole-genome sequencing.
© 2023 The Authors. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
NR and DG are co‐founders of Inivata. CGS is a current employee of Inivata. DG, JAM, and KH are current employees of AstraZeneca. Inivata and AstraZeneca had no role in the conceptualization or design of the clinical study, statistical analysis, or decision to publish the manuscript. All other authors declare that they have no competing interests.
Figures

References
-
- Almogy G, Pratt M, Oberstrass F, Lee L, Mazur D, Beckett N, Barad O, Soifer I, Perelman E, Etzioni Y et al (2022) Cost‐efficient whole genome‐sequencing using novel mostly natural sequencing‐by‐synthesis chemistry and open fluidics platform. bioRxiv 10.1101/2022.05.29.493900 [PREPRINT] - DOI
-
- Broad Institute (2019) Picard Toolkit. http://broadinstitute.github.io/picard/
Publication types
MeSH terms
Substances
Grants and funding
- 337905/ERC_/European Research Council/International
- A20240/CRUK_/Cancer Research UK/United Kingdom
- A21491/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_00002/16/MRC_/Medical Research Council/United Kingdom
- C507/A27657/CRUK_/Cancer Research UK/United Kingdom
- C36857/A27548/CRUK_/Cancer Research UK/United Kingdom
- C9545/A29580/CRUK_/Cancer Research UK/United Kingdom
- 21491/CRUK_/Cancer Research UK/United Kingdom
- C1287/A26886/CRUK_/Cancer Research UK/United Kingdom
- BRC-1215-20014/DH_/Department of Health/United Kingdom
- 25117/CRUK_/Cancer Research UK/United Kingdom
- C17918/A28870/CRUK_/Cancer Research UK/United Kingdom
- A28870/CRUK_/Cancer Research UK/United Kingdom
- C9685/A25177/CRUK_/Cancer Research UK/United Kingdom
- 106566/WT_/Wellcome Trust/United Kingdom
- 20240/CRUK_/Cancer Research UK/United Kingdom
- EDDCPT\100013/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
