

Establishing Quality Control Procedures for Large-Scale Plasma Proteomics Analyses
- PMID: 37163770
- PMCID: PMC12204224
- DOI: 10.1021/jasms.3c00050
Establishing Quality Control Procedures for Large-Scale Plasma Proteomics Analyses
Abstract
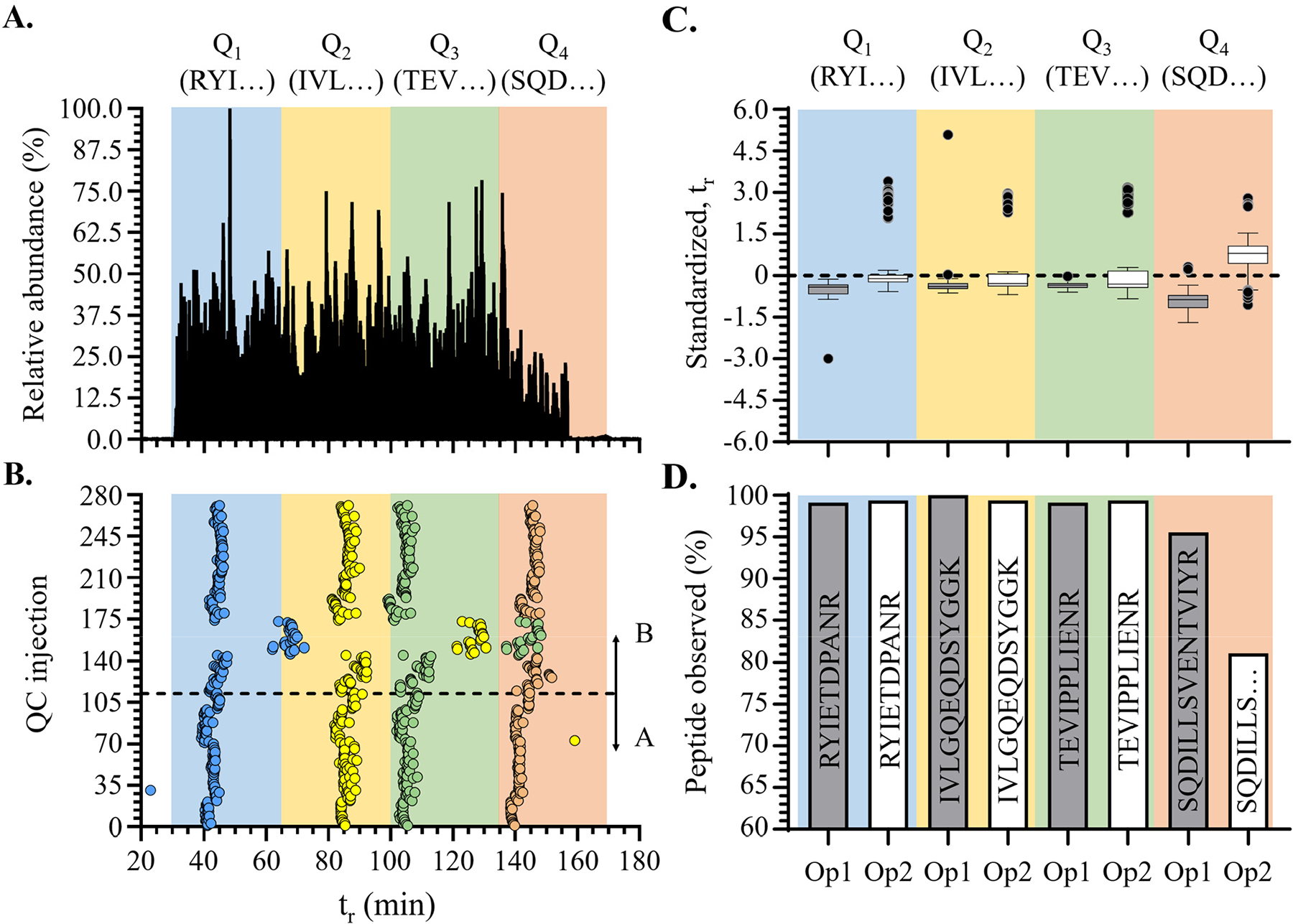
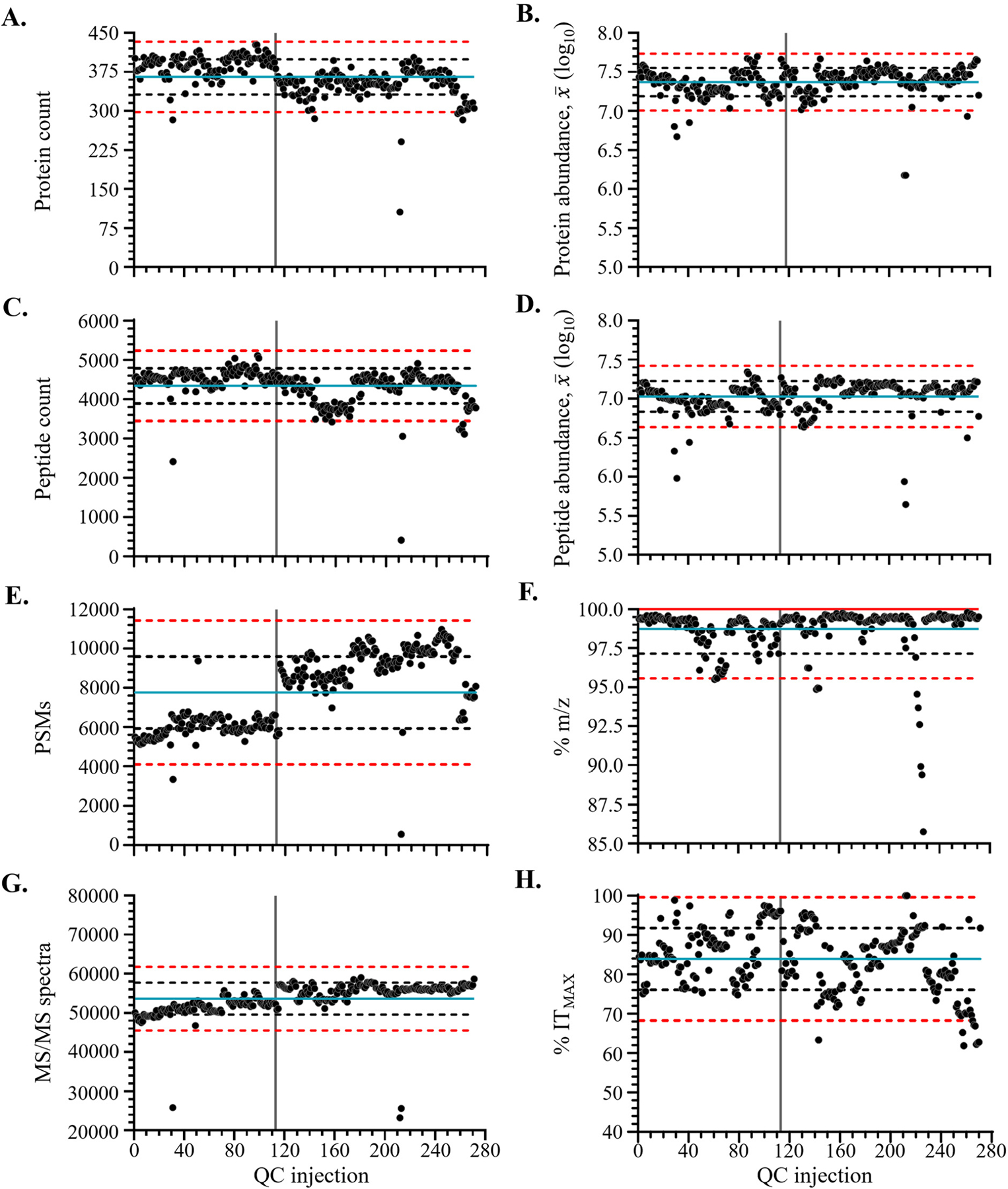
Proteomics research has been transformed due to high-throughput liquid chromatography (LC-MS/MS) tandem mass spectrometry instruments combined with highly sophisticated automated sample preparation and multiplexing workflows. However, scaling proteomics experiments to large sample cohorts (hundreds to thousands) requires thoughtful quality control (QC) protocols. Robust QC protocols can help with reproducibility, quantitative accuracy, and provide opportunities for more decisive troubleshooting. Our laboratory conducted a plasma proteomics study of a cohort of N = 335 patient samples using tandem mass tag (TMTpro) 16-plex batches. Over the course of a 10-month data acquisition period for this cohort we collected 271 pooled QC LC-MS/MS result files obtained from MS/MS analysis of a patient-derived pooled plasma sample, representative of the entire cohort population. This sample was tagged with TMTzero or TMTpro reagents and used to inform the daily performance of the LC-MS/MS instruments and to allow within and across sample batch normalization. Analytical variability of a number of instrumental and data analysis metrics including protein and peptide identifications, peptide spectral matches (PSMs), number of obtained MS/MS spectra, average peptide abundance, percent of peptides with a Δ m/z between ±0.003 Da, percent of MS/MS spectra obtained at the maximum injection time, and the retention time of selected tracking peptides were evaluated to help inform the design of a robust LC-MS/MS QC workflow for use in future cohort studies. This study also led to general tips for using selected metrics to inform real-time troubleshooting of LC-MS/MS performance issues with daily QC checks.
Keywords: liquid chromatography; mass spectrometry; plasma; proteins; proteomics; quality control.
Conflict of interest statement
The authors declare no competing financial interest.
Figures



References
-
- Dubois C; Payen D; Simon S; Junot C; Fenaille F; Morel N; Becher F Top-Down and Bottom-Up Proteomics of Circulating S100A8/S100A9 in Plasma of Septic Shock Patients. J. Proteome Res 2020, 19 (2), 914–925. - PubMed
-
- Solimani F; Didona D; Li J; Bao L; Patel PM; Gasparini G; Kridin K; Cozzani E; Hertl M; Amber KT Characterizing the proteome of bullous pemphigoid blister fluid utilizing tandem mass tag labeling coupled with LC-MS/MS. Arch Dermatol Res. 2022, 314 (9), 921–928. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
