

Gaps and complex structurally variant loci in phased genome assemblies
- PMID: 37164484
- PMCID: PMC10234299
- DOI: 10.1101/gr.277334.122
Gaps and complex structurally variant loci in phased genome assemblies
Abstract
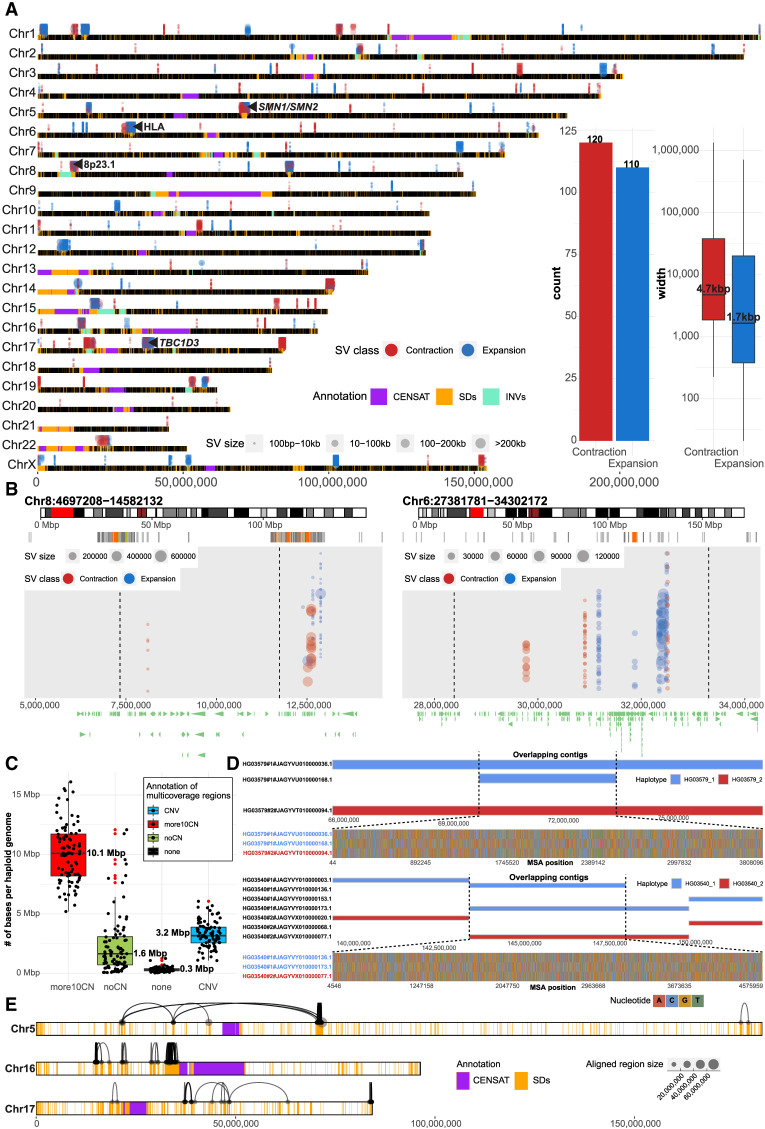
There has been tremendous progress in phased genome assembly production by combining long-read data with parental information or linked-read data. Nevertheless, a typical phased genome assembly generated by trio-hifiasm still generates more than 140 gaps. We perform a detailed analysis of gaps, assembly breaks, and misorientations from 182 haploid assemblies obtained from a diversity panel of 77 unique human samples. Although trio-based approaches using HiFi are the current gold standard, chromosome-wide phasing accuracy is comparable when using Strand-seq instead of parental data. Importantly, the majority of assembly gaps cluster near the largest and most identical repeats (including segmental duplications [35.4%], satellite DNA [22.3%], or regions enriched in GA/AT-rich DNA [27.4%]). Consequently, 1513 protein-coding genes overlap assembly gaps in at least one haplotype, and 231 are recurrently disrupted or missing from five or more haplotypes. Furthermore, we estimate that 6-7 Mbp of DNA are misorientated per haplotype irrespective of whether trio-free or trio-based approaches are used. Of these misorientations, 81% correspond to bona fide large inversion polymorphisms in the human species, most of which are flanked by large segmental duplications. We also identify large-scale alignment discontinuities consistent with 11.9 Mbp of deletions and 161.4 Mbp of insertions per haploid genome. Although 99% of this variation corresponds to satellite DNA, we identify 230 regions of euchromatic DNA with frequent expansions and contractions, nearly half of which overlap with 197 protein-coding genes. Such variable and incompletely assembled regions are important targets for future algorithmic development and pangenome representation.
© 2023 Porubsky et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
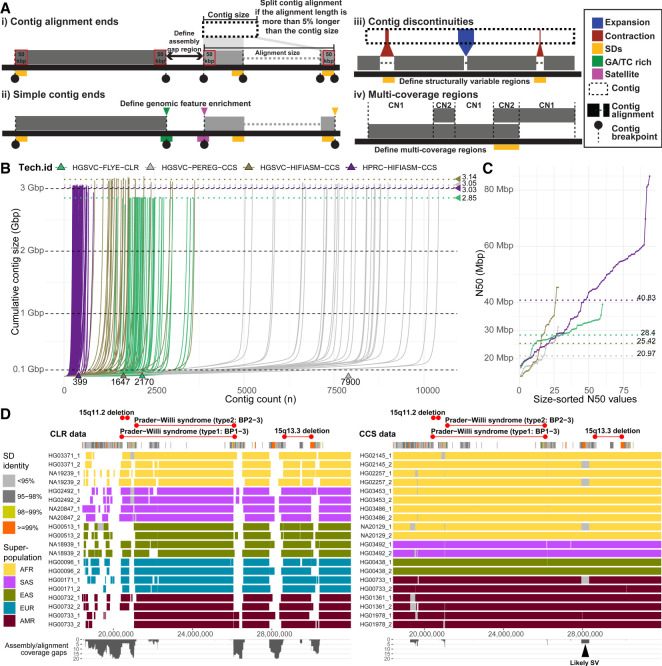
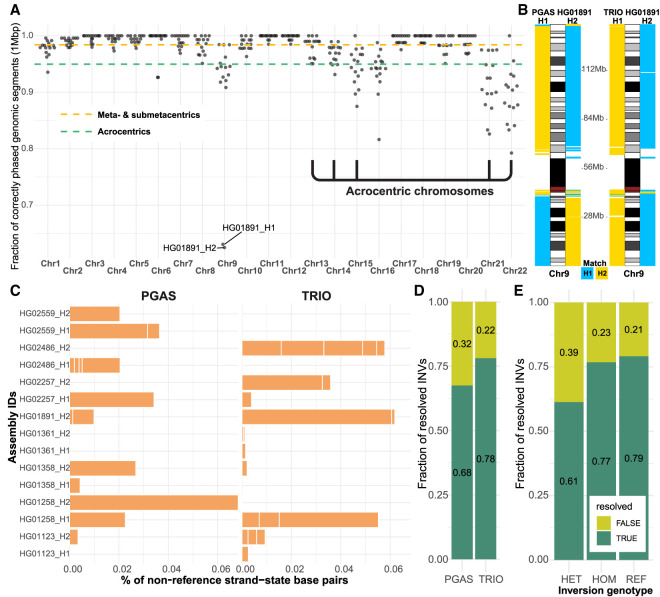
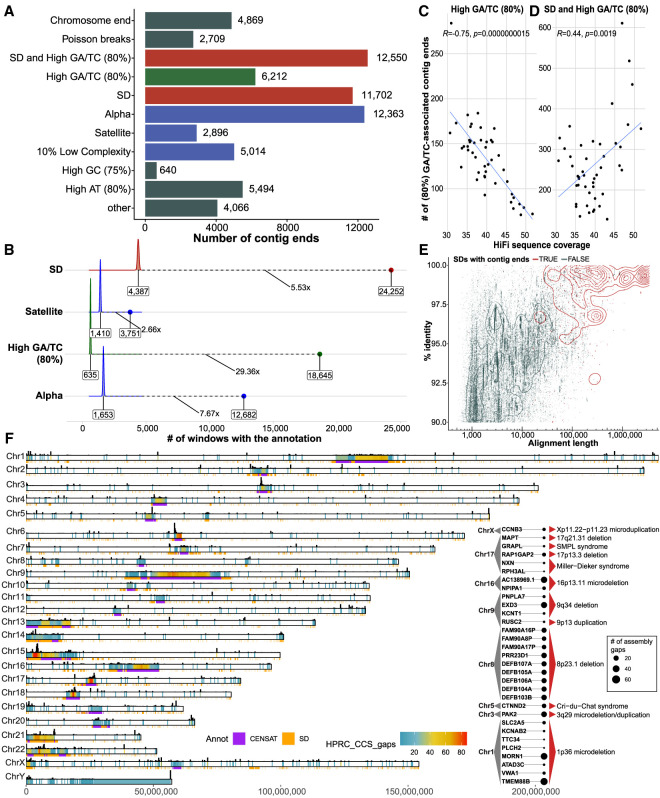
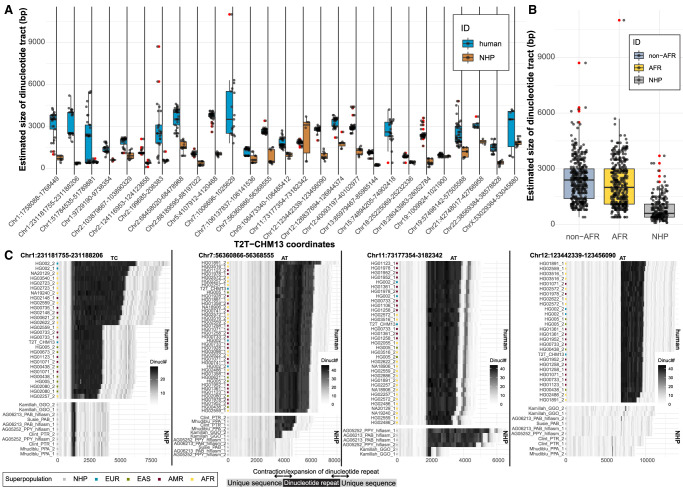
References
-
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. 2022. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185: 3426–3440.e19. 10.1016/j.cell.2022.08.004 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous