

Pangenome graph construction from genome alignments with Minigraph-Cactus
- PMID: 37165083
- PMCID: PMC10638906
- DOI: 10.1038/s41587-023-01793-w
Pangenome graph construction from genome alignments with Minigraph-Cactus
Abstract
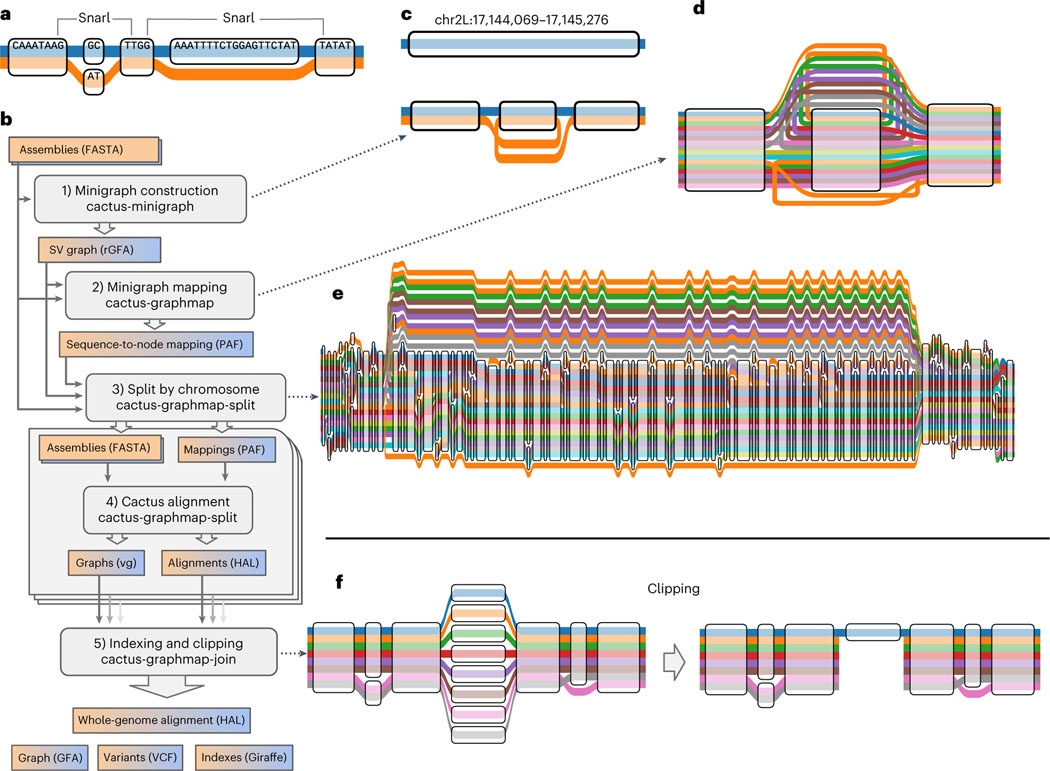
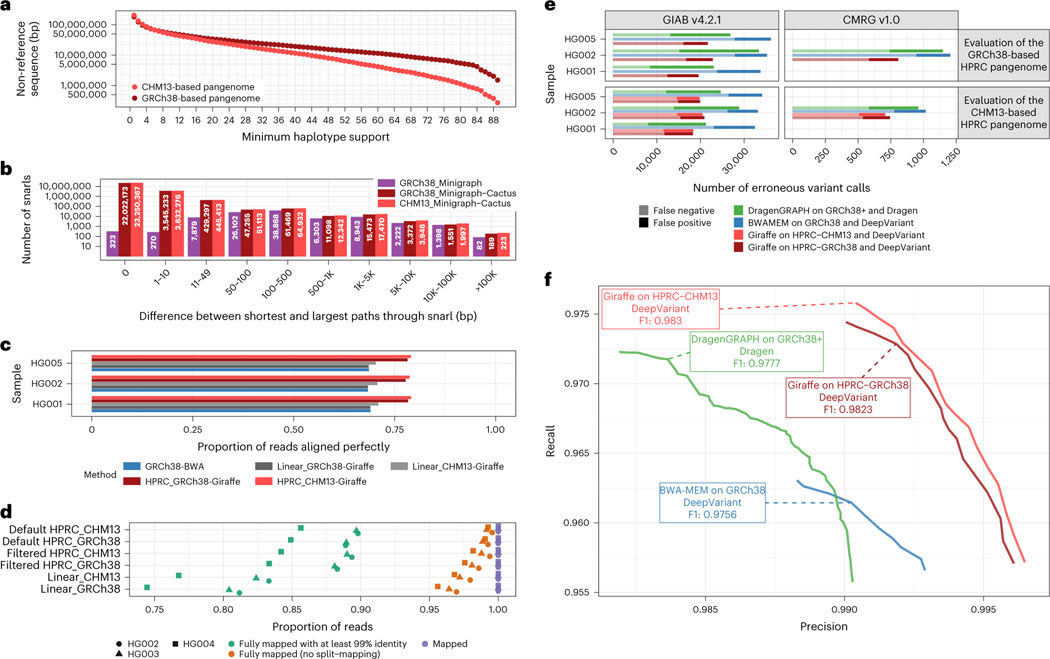
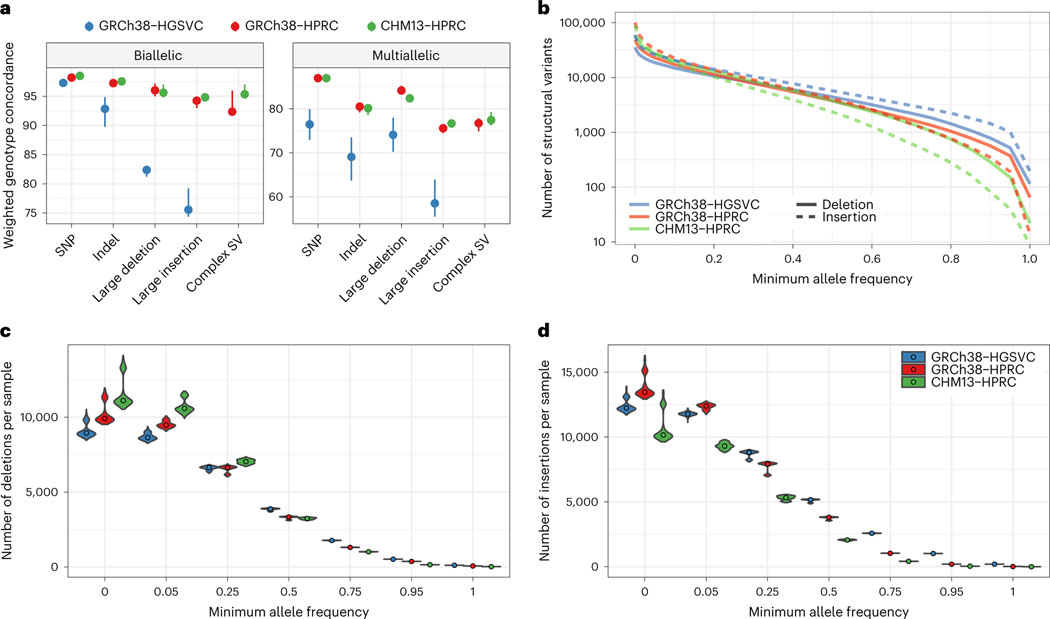
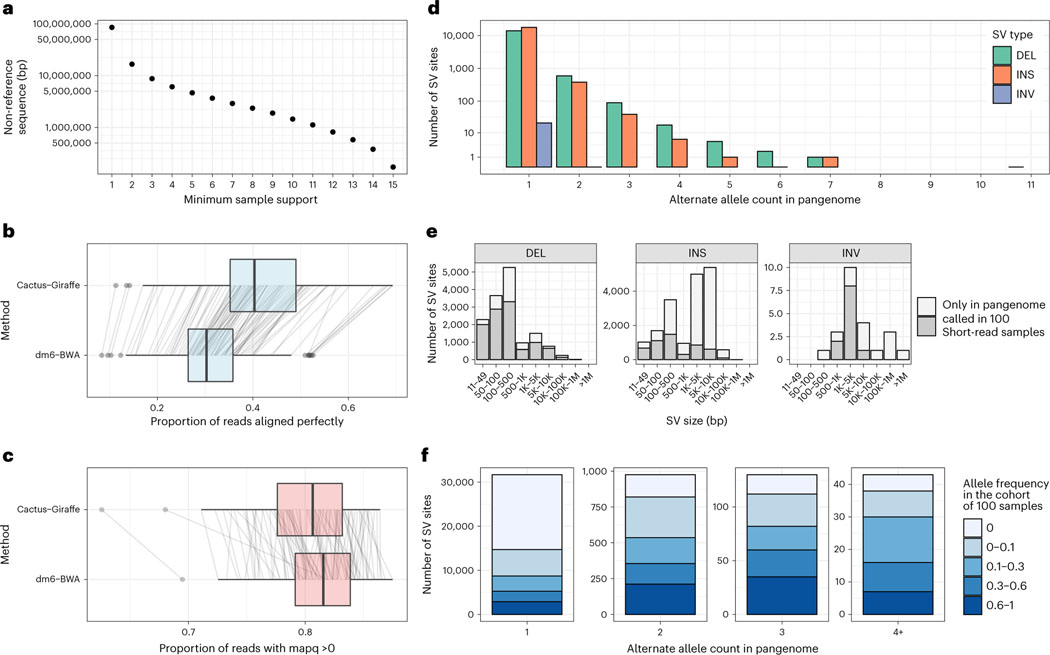
Pangenome references address biases of reference genomes by storing a representative set of diverse haplotypes and their alignment, usually as a graph. Alternate alleles determined by variant callers can be used to construct pangenome graphs, but advances in long-read sequencing are leading to widely available, high-quality phased assemblies. Constructing a pangenome graph directly from assemblies, as opposed to variant calls, leverages the graph's ability to represent variation at different scales. Here we present the Minigraph-Cactus pangenome pipeline, which creates pangenomes directly from whole-genome alignments, and demonstrate its ability to scale to 90 human haplotypes from the Human Pangenome Reference Consortium. The method builds graphs containing all forms of genetic variation while allowing use of current mapping and genotyping tools. We measure the effect of the quality and completeness of reference genomes used for analysis within the pangenomes and show that using the CHM13 reference from the Telomere-to-Telomere Consortium improves the accuracy of our methods. We also demonstrate construction of a Drosophila melanogaster pangenome.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
