

Profiling the human intestinal environment under physiological conditions
- PMID: 37165188
- PMCID: PMC10191855
- DOI: 10.1038/s41586-023-05989-7
Profiling the human intestinal environment under physiological conditions
Abstract
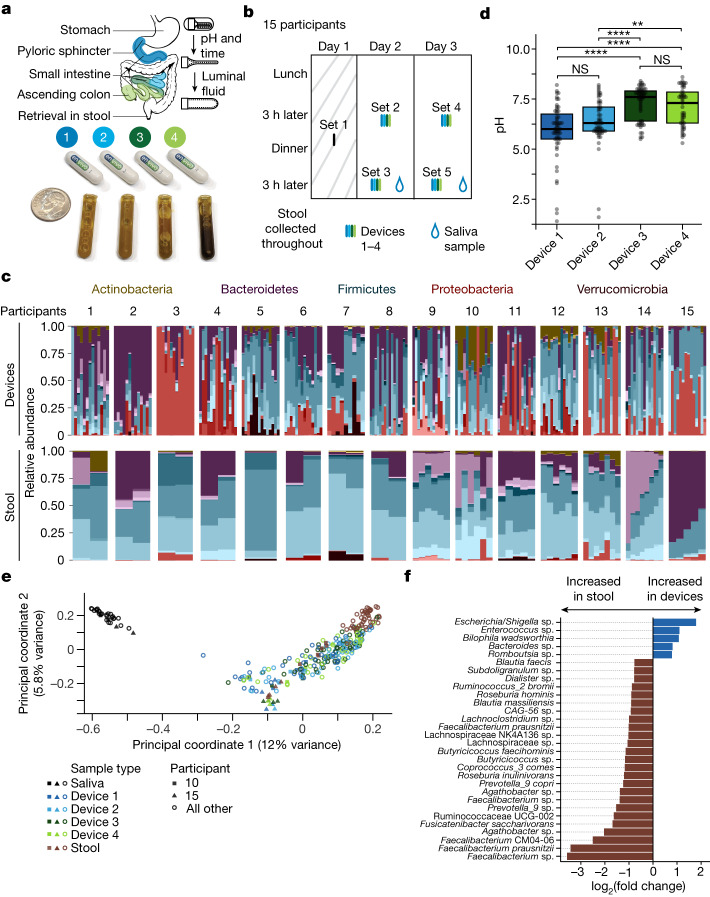
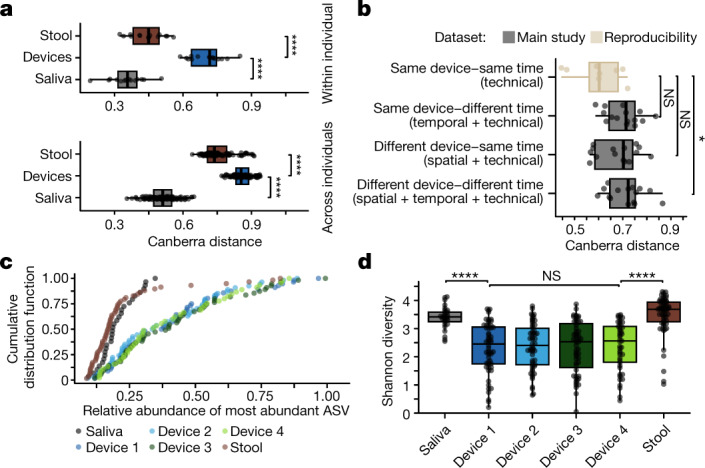
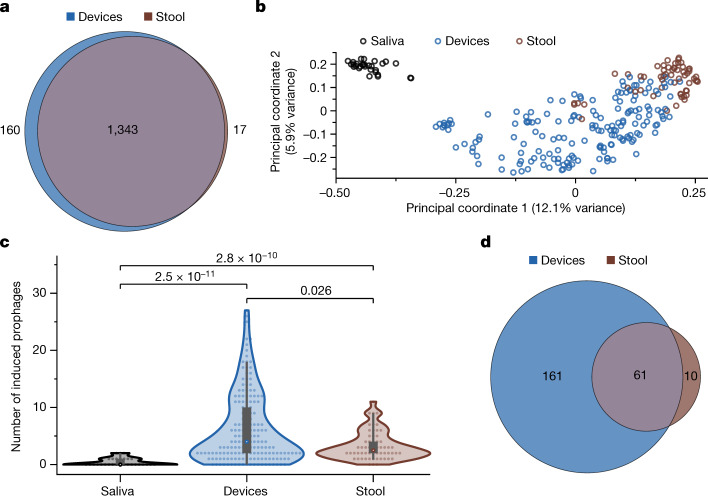
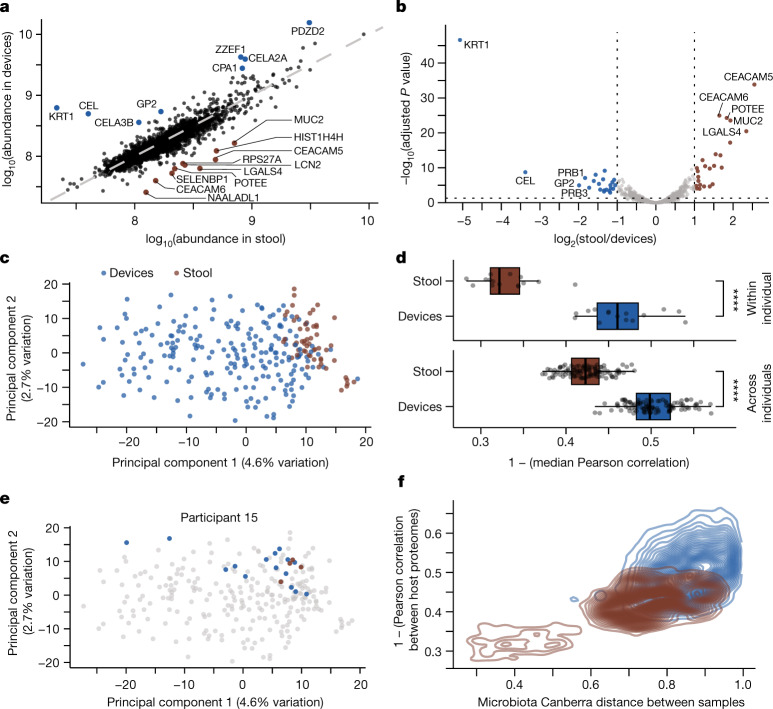
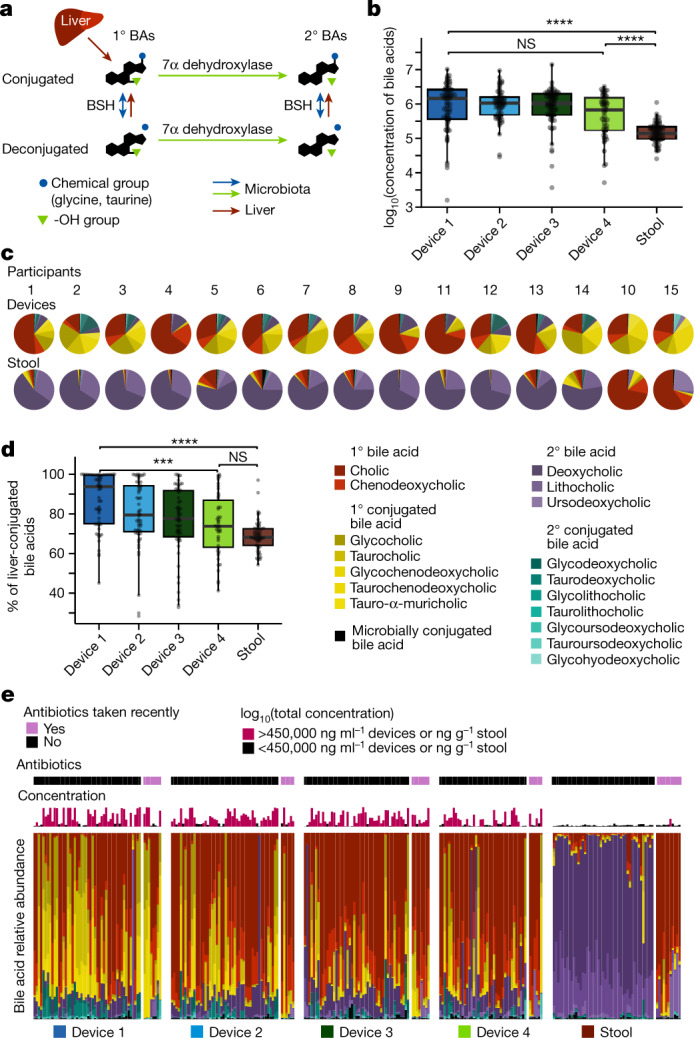
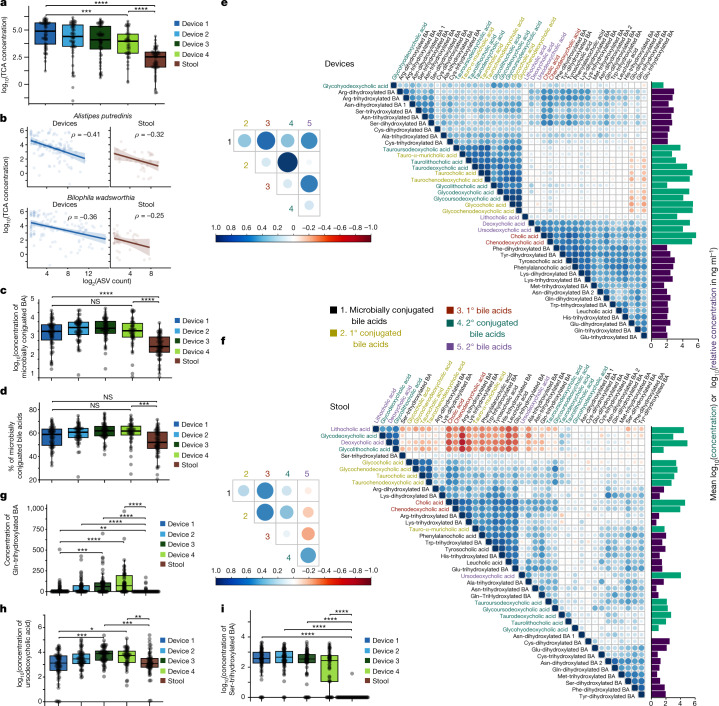
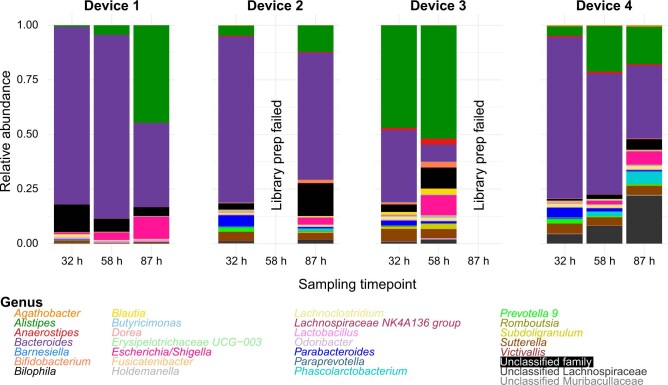
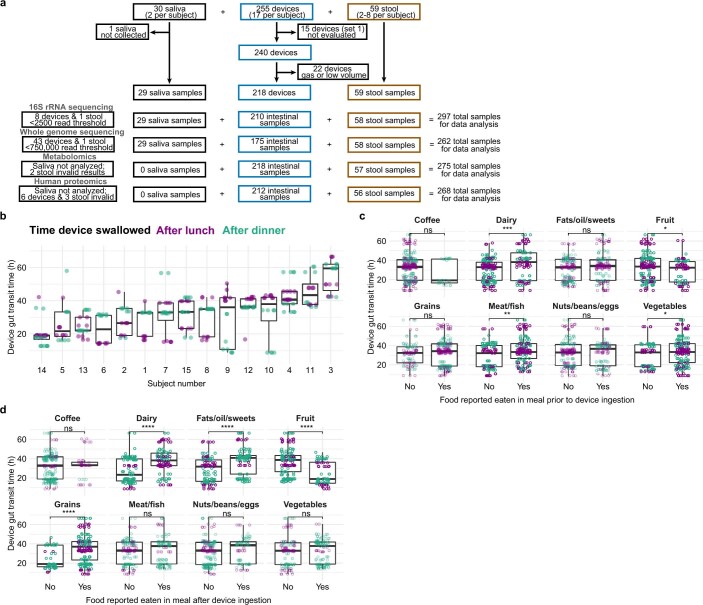
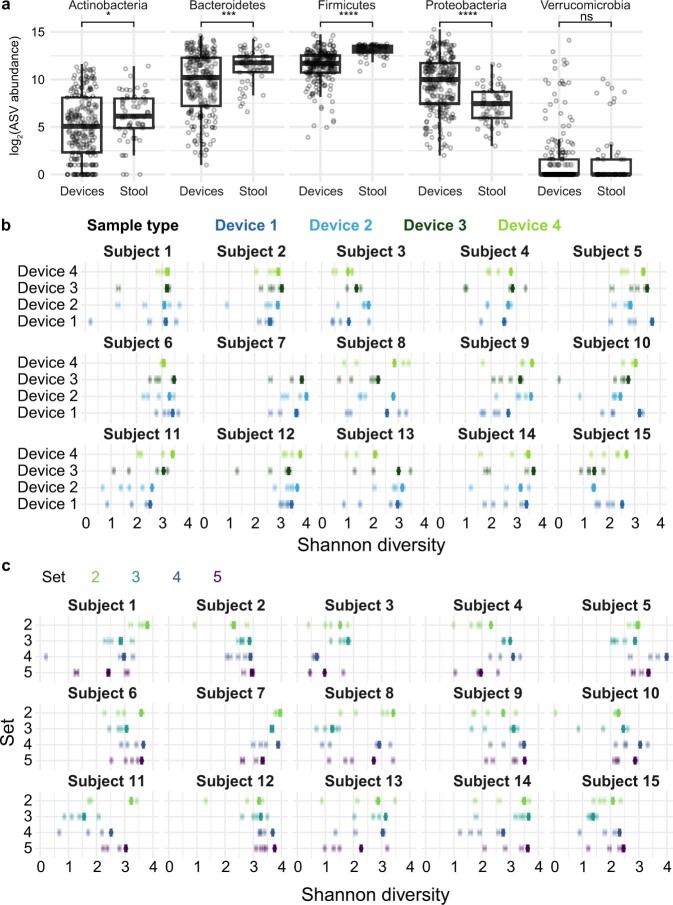
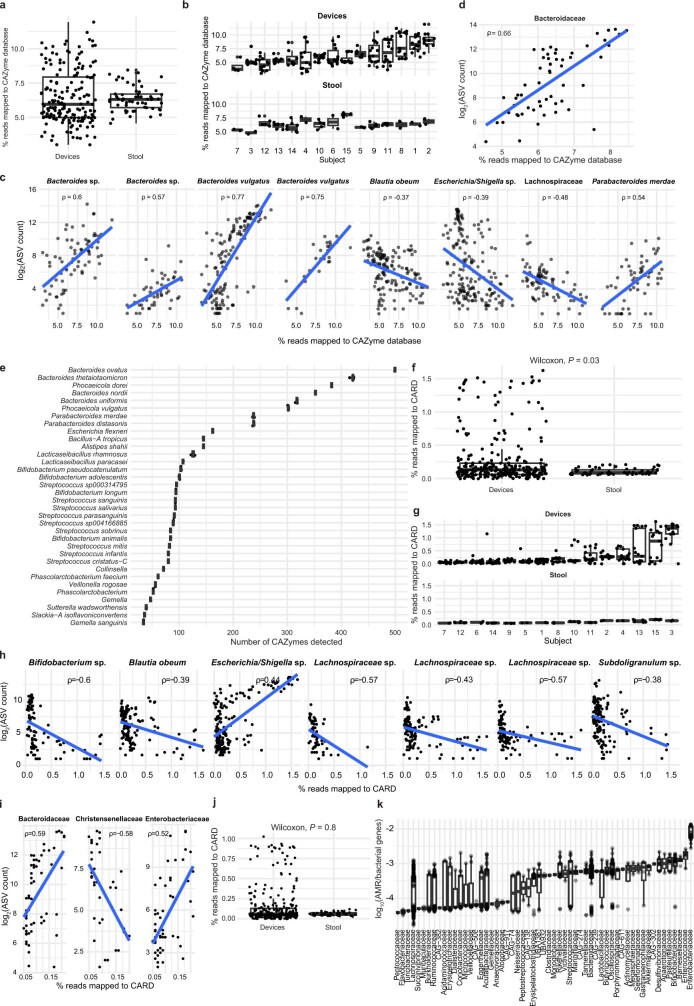
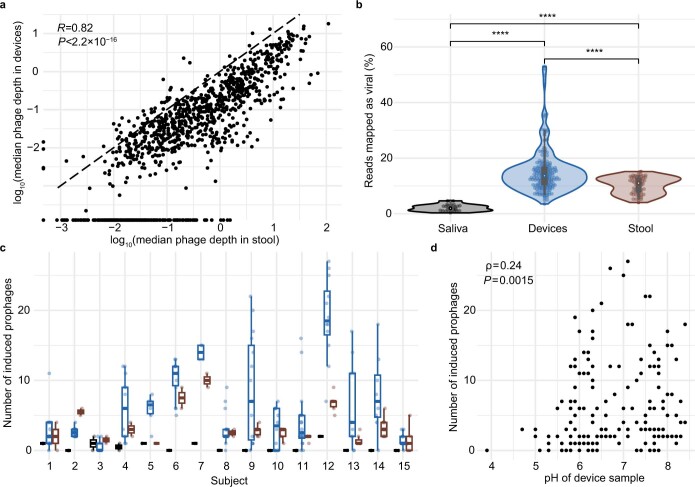
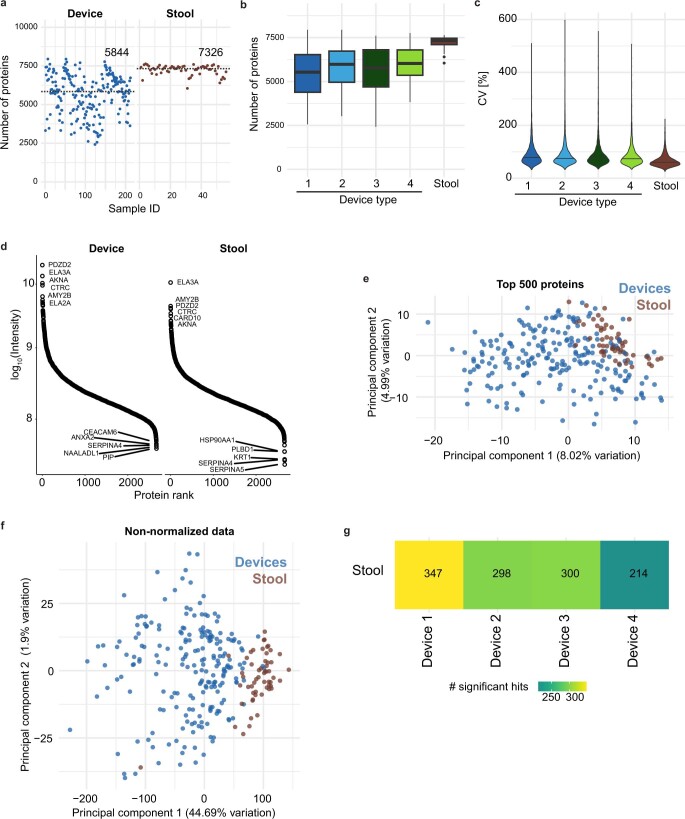
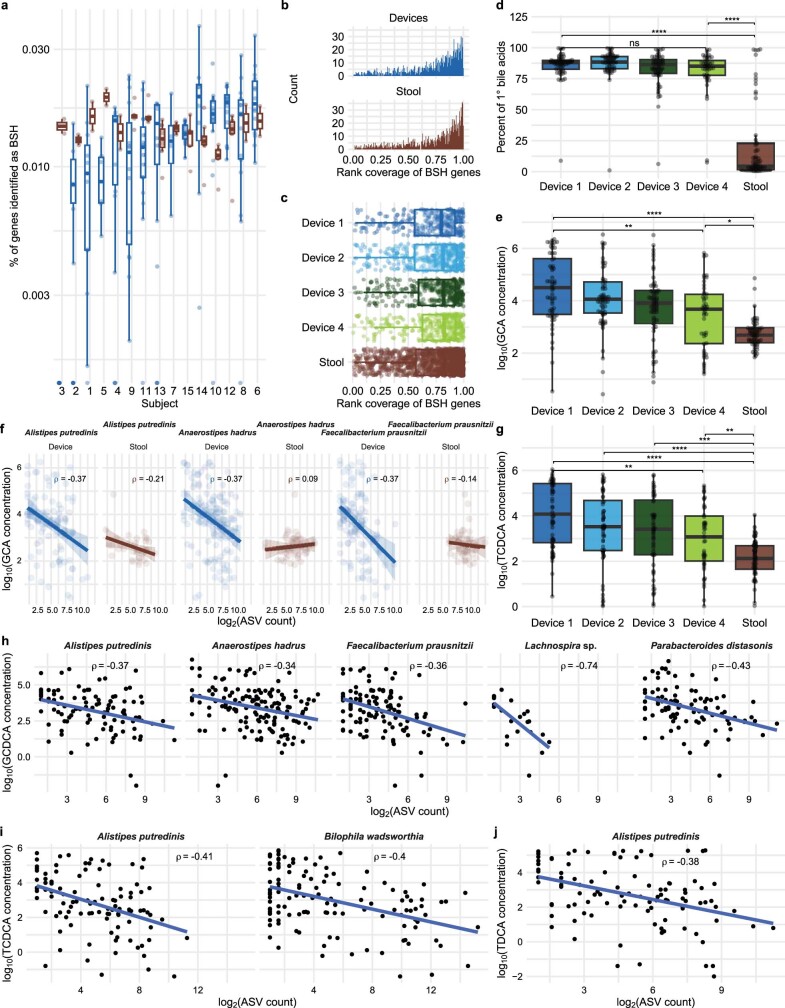
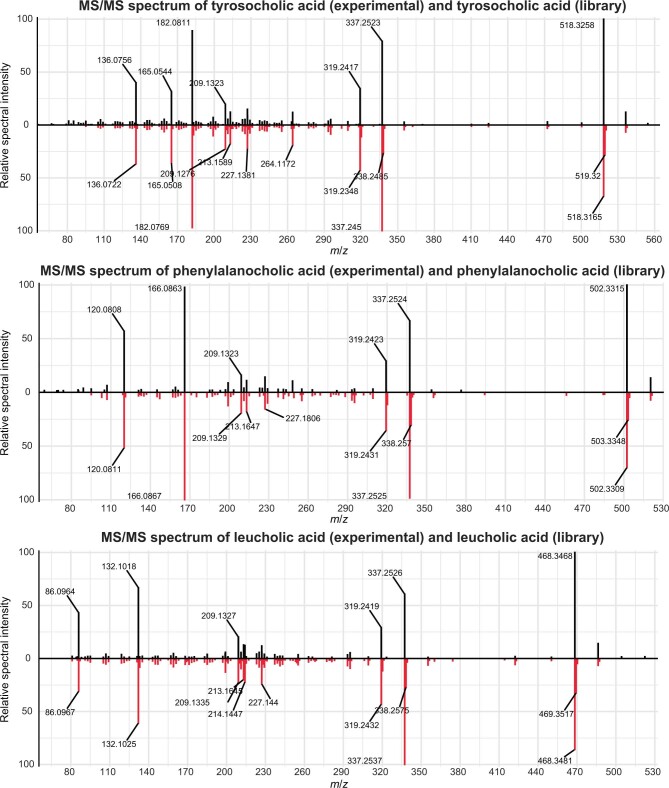
The spatiotemporal structure of the human microbiome1,2, proteome3 and metabolome4,5 reflects and determines regional intestinal physiology and may have implications for disease6. Yet, little is known about the distribution of microorganisms, their environment and their biochemical activity in the gut because of reliance on stool samples and limited access to only some regions of the gut using endoscopy in fasting or sedated individuals7. To address these deficiencies, we developed an ingestible device that collects samples from multiple regions of the human intestinal tract during normal digestion. Collection of 240 intestinal samples from 15 healthy individuals using the device and subsequent multi-omics analyses identified significant differences between bacteria, phages, host proteins and metabolites in the intestines versus stool. Certain microbial taxa were differentially enriched and prophage induction was more prevalent in the intestines than in stool. The host proteome and bile acid profiles varied along the intestines and were highly distinct from those of stool. Correlations between gradients in bile acid concentrations and microbial abundance predicted species that altered the bile acid pool through deconjugation. Furthermore, microbially conjugated bile acid concentrations exhibited amino acid-dependent trends that were not apparent in stool. Overall, non-invasive, longitudinal profiling of microorganisms, proteins and bile acids along the intestinal tract under physiological conditions can help elucidate the roles of the gut microbiome and metabolome in human physiology and disease.
© 2023. The Author(s).
Conflict of interest statement
D.S. is an employee of Envivo Bio, Inc. (San Francisco, CA) and owns stock in the company. D.S. is an inventor on pending patent application WO2018213729 covering the sampling device described in the manuscript, which is owned by Envivo Bio, Inc. All other authors declare no competing interests.
Figures
Comment in
-
A multi-omic trip through the human gut.Nat Metab. 2023 May;5(5):720-721. doi: 10.1038/s42255-023-00773-3. Nat Metab. 2023. PMID: 37165175 No abstract available.
