

A pan-grass transcriptome reveals patterns of cellular divergence in crops
- PMID: 37165193
- PMCID: PMC10657638
- DOI: 10.1038/s41586-023-06053-0
A pan-grass transcriptome reveals patterns of cellular divergence in crops
Abstract
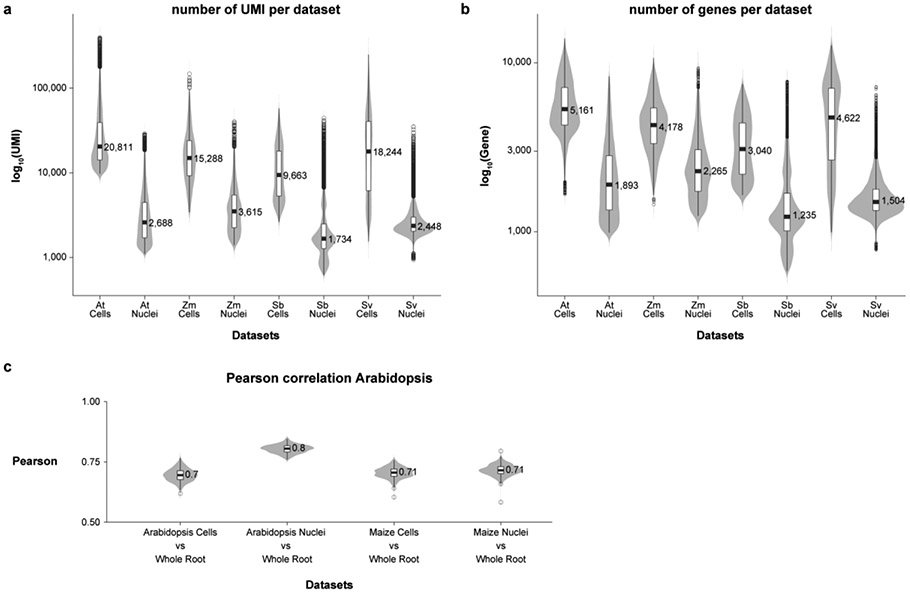
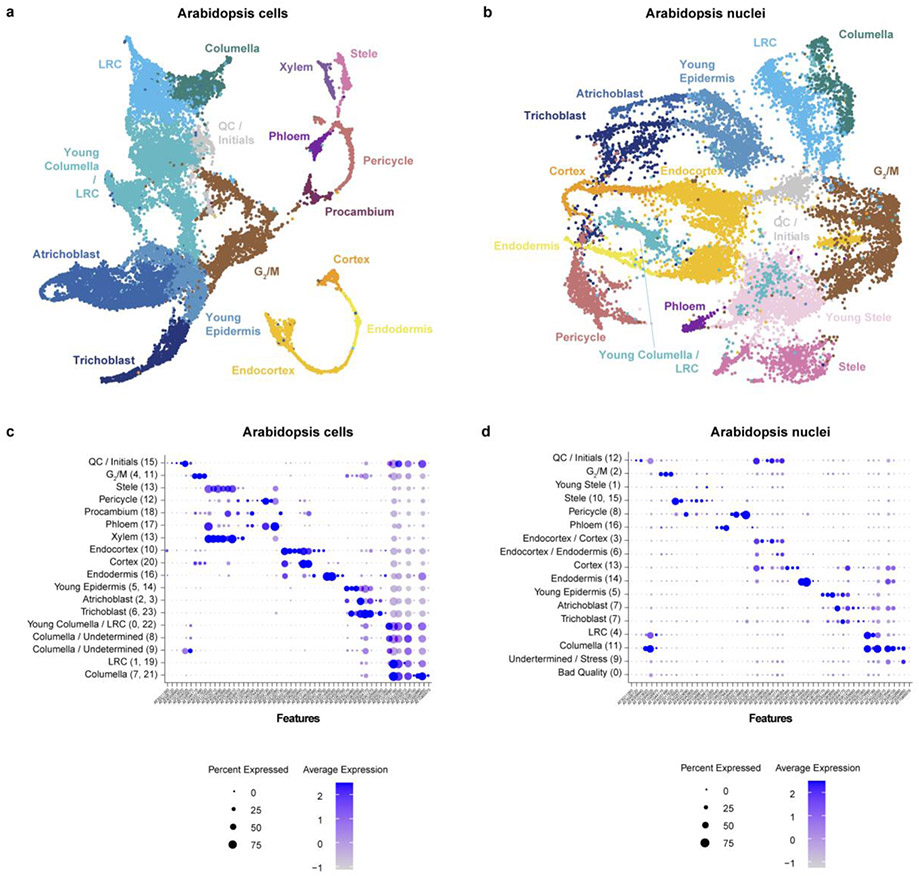
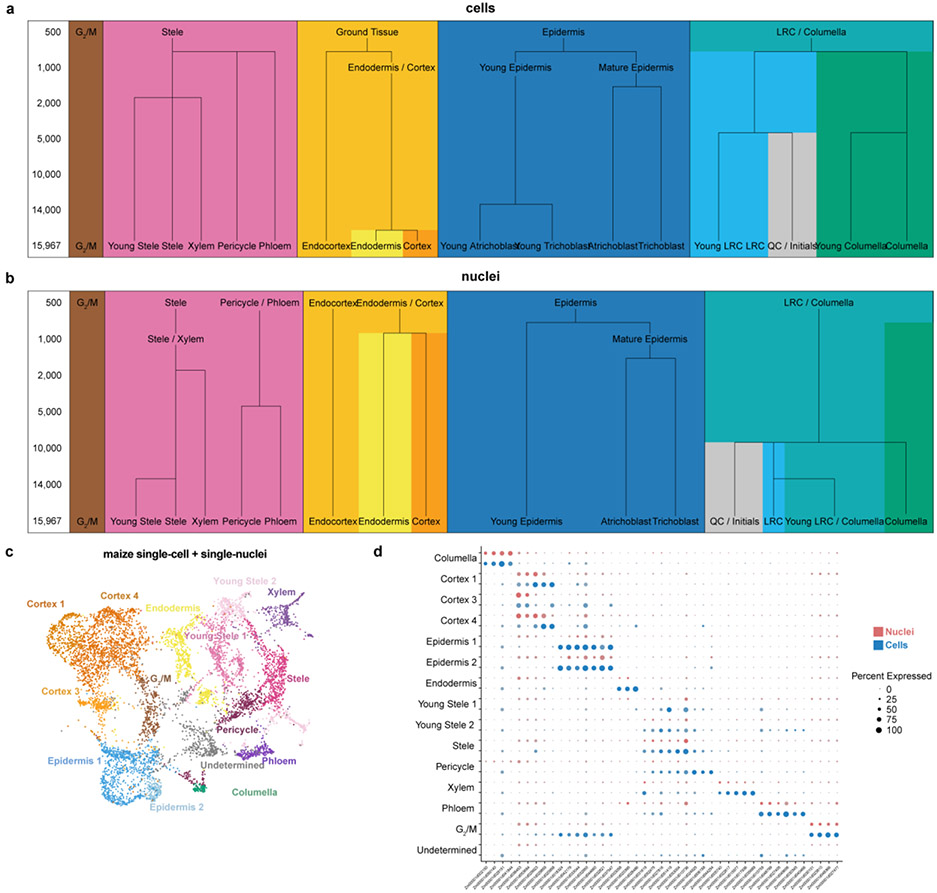
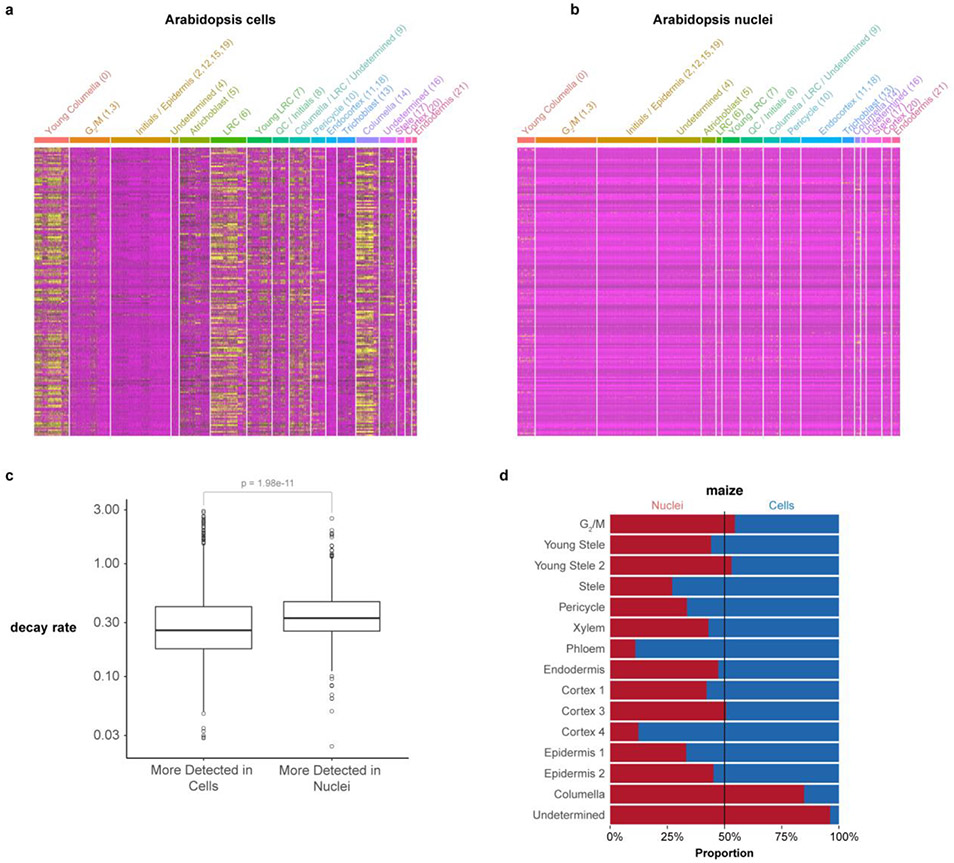
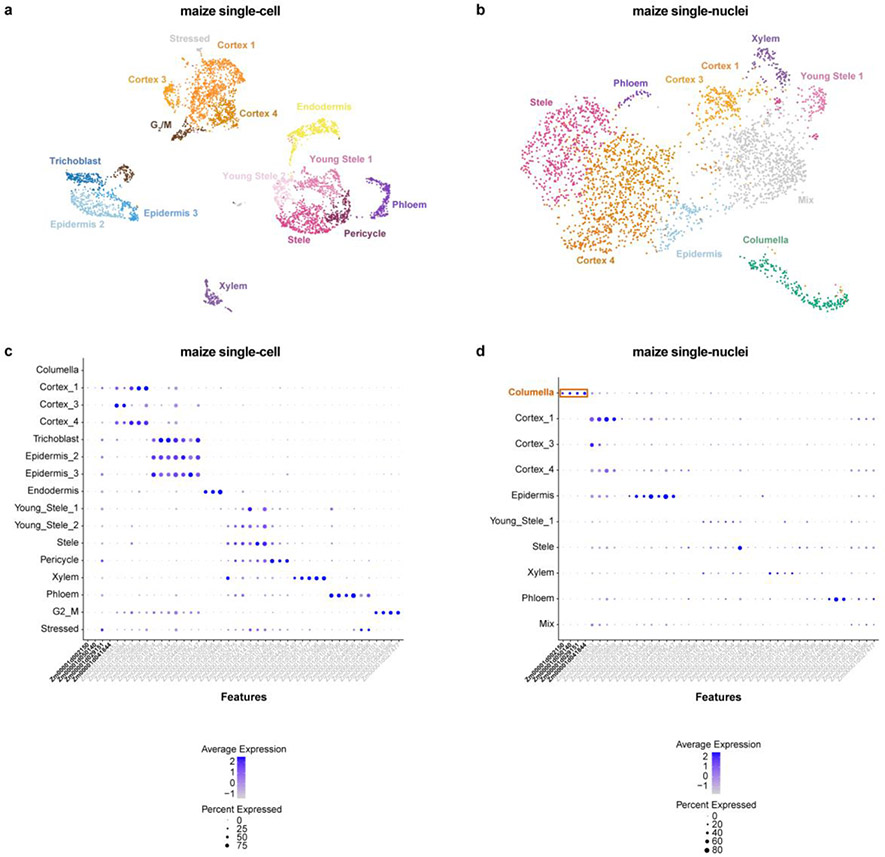
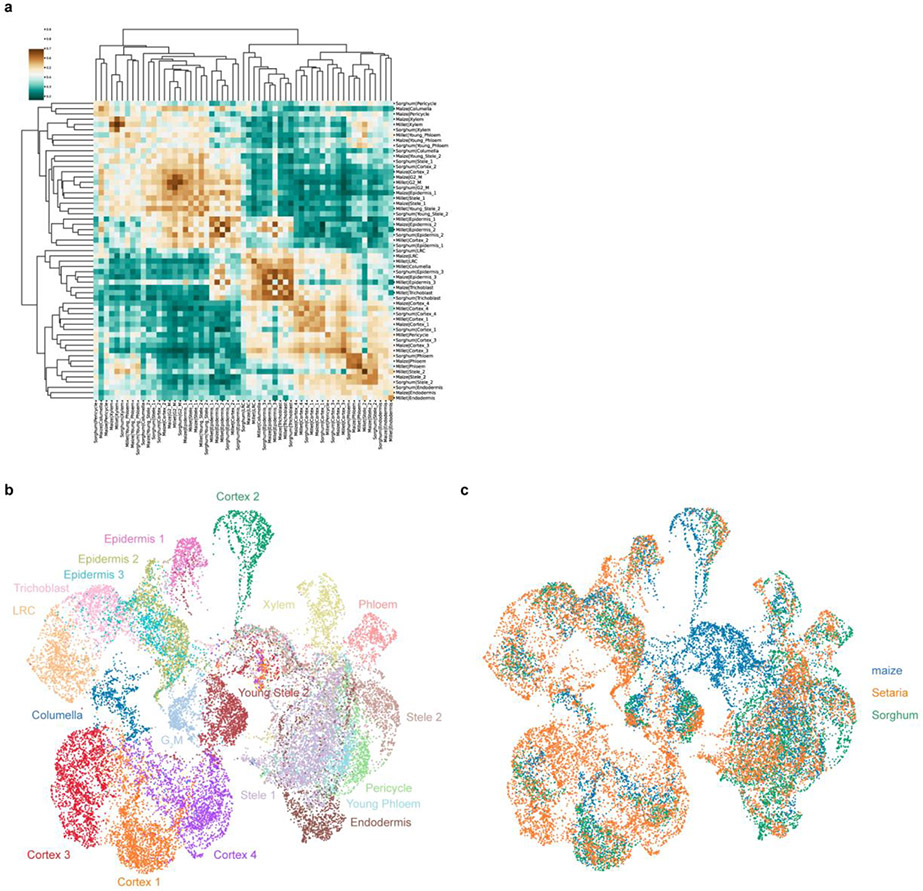
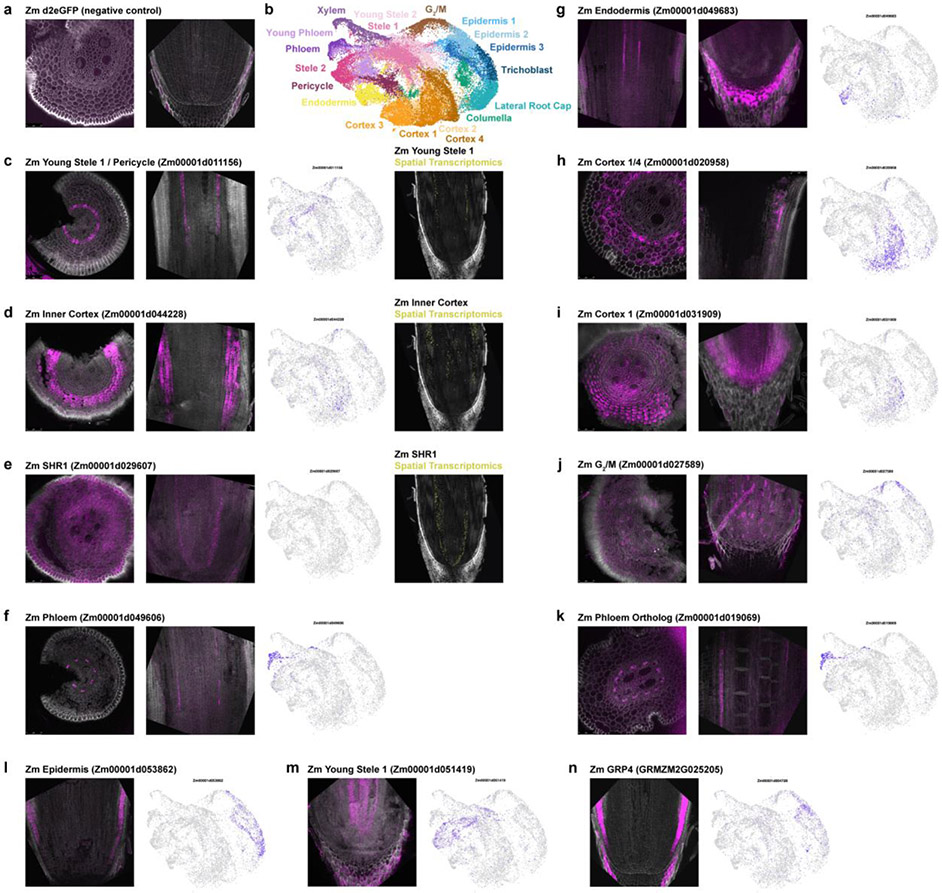
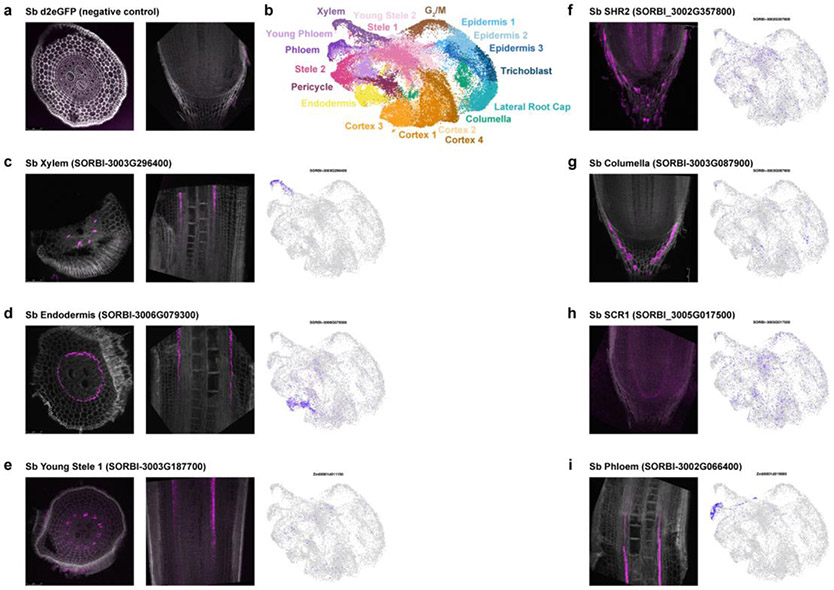
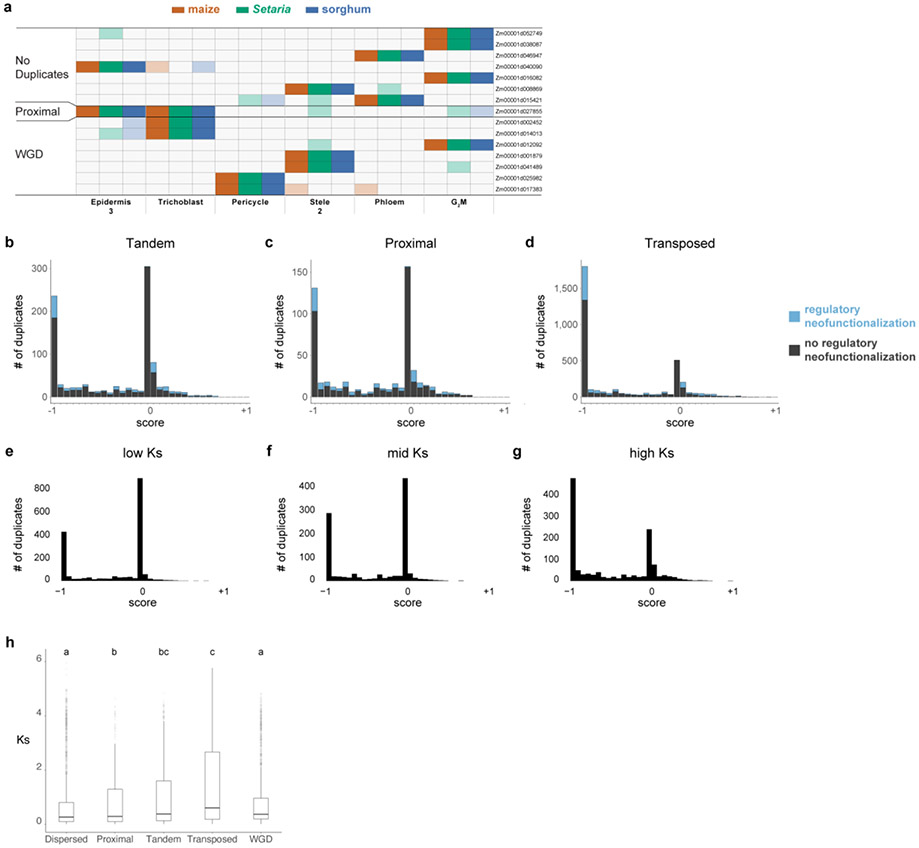
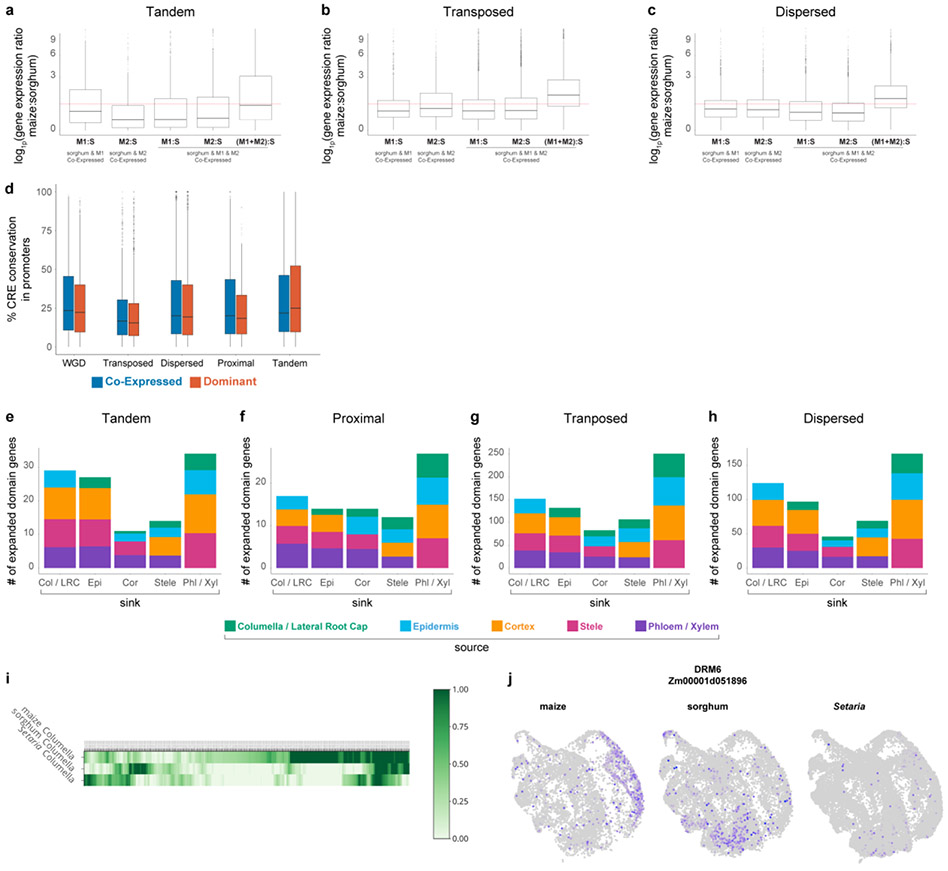
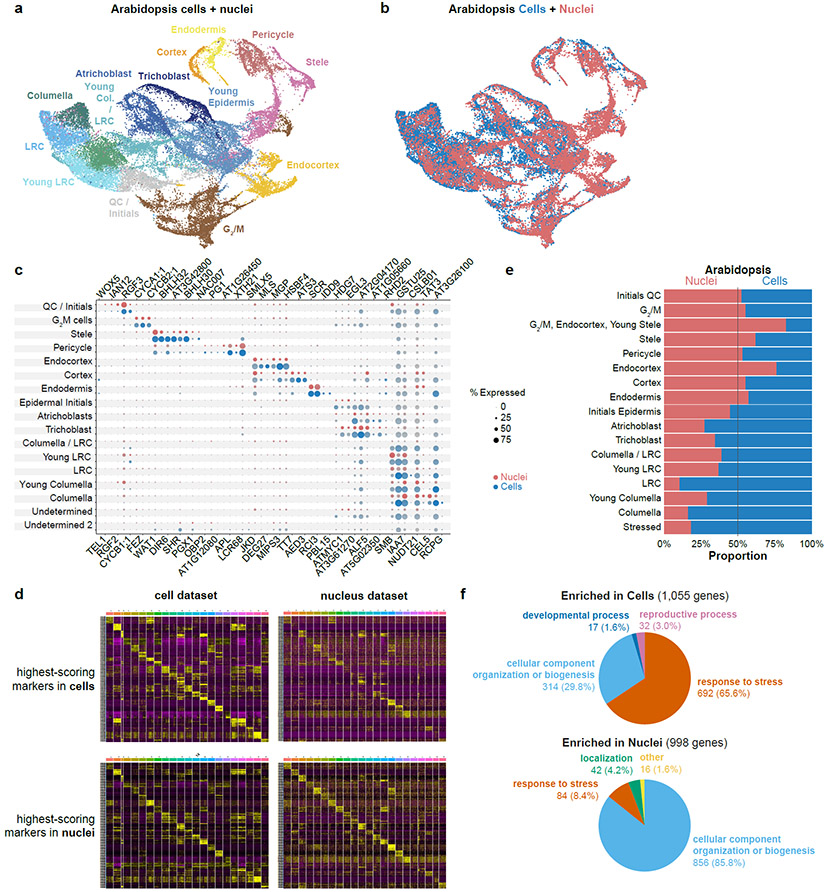
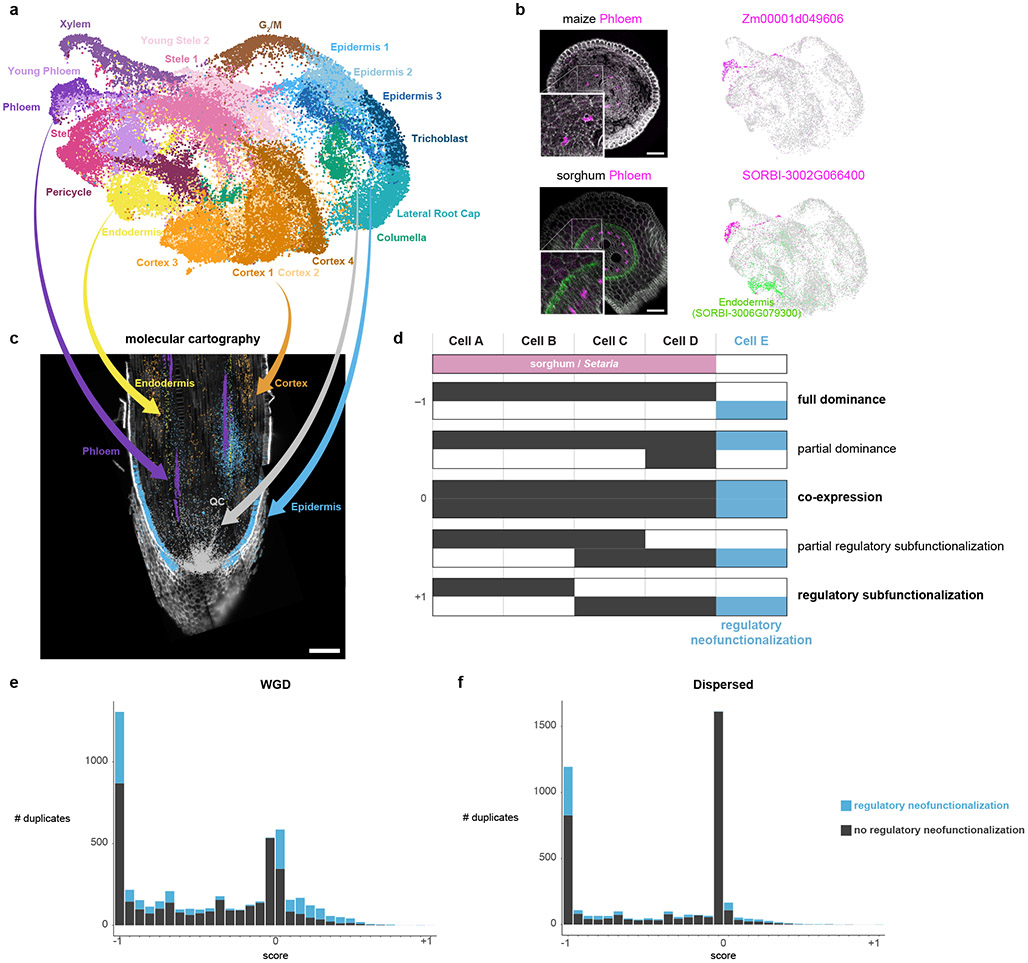
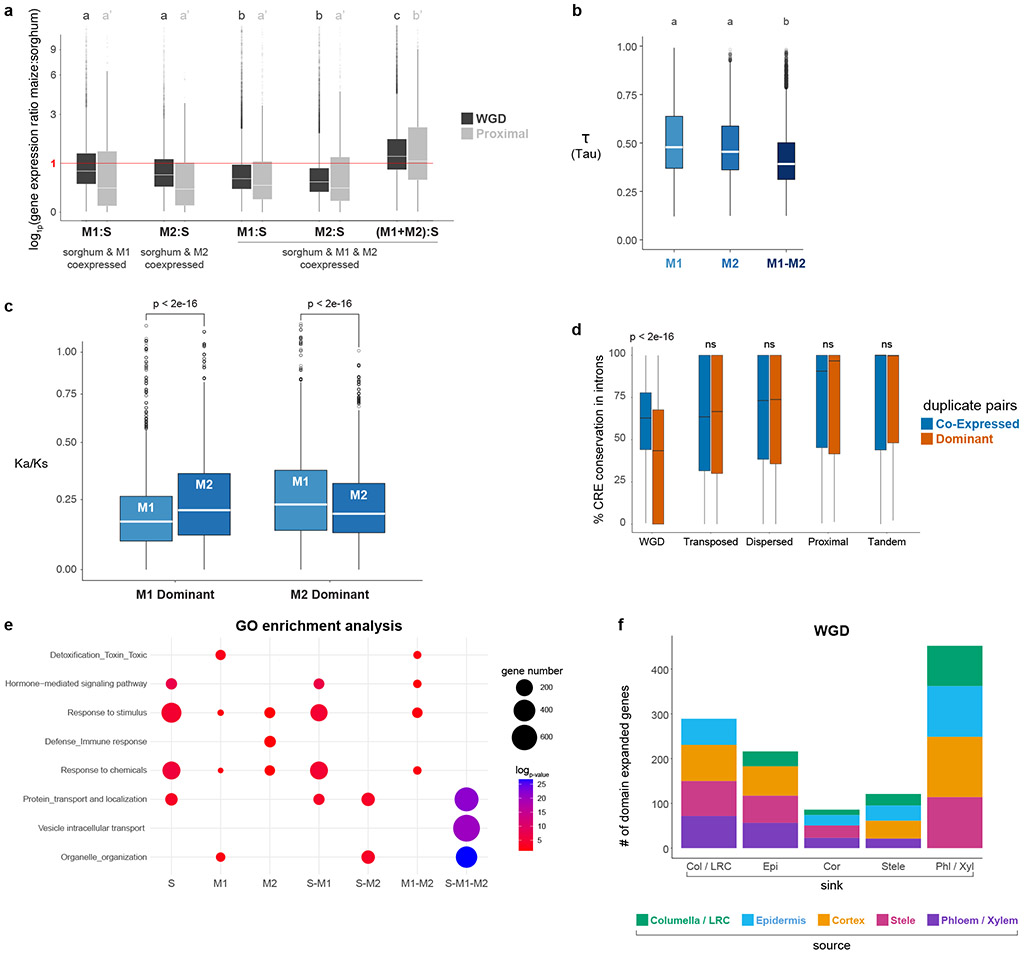
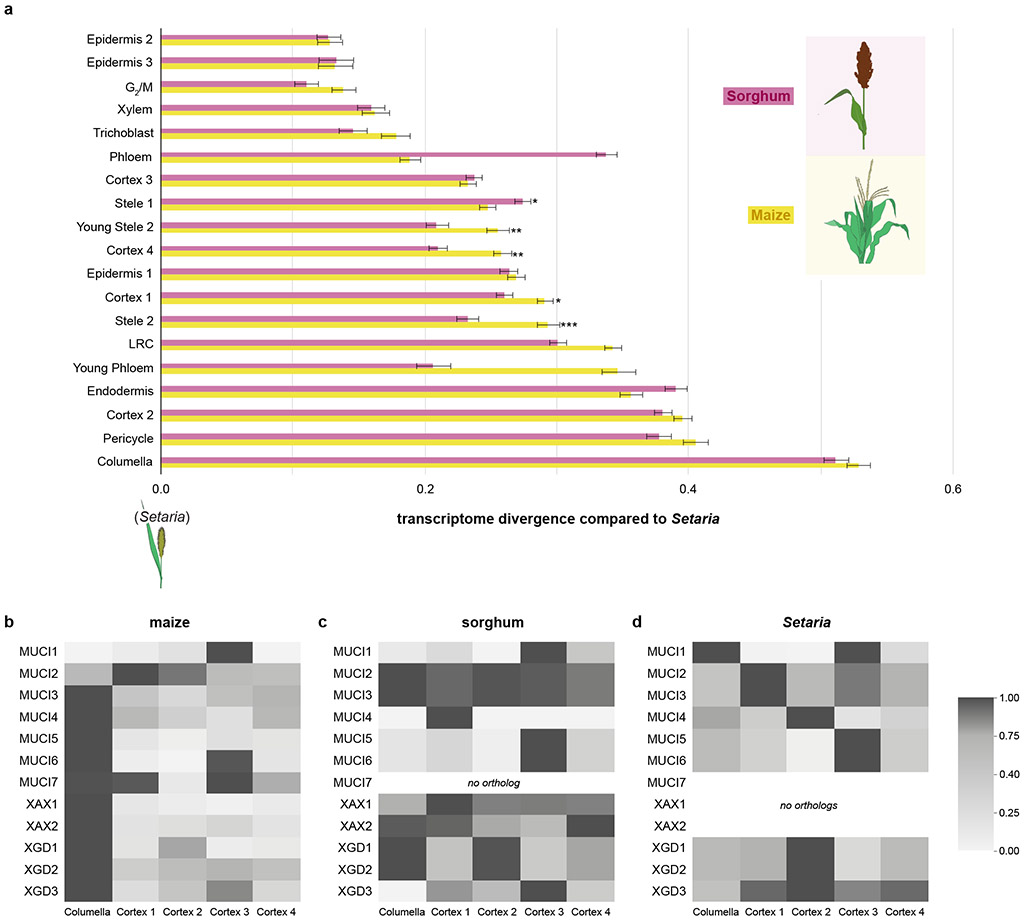
Different plant species within the grasses were parallel targets of domestication, giving rise to crops with distinct evolutionary histories and traits1. Key traits that distinguish these species are mediated by specialized cell types2. Here we compare the transcriptomes of root cells in three grass species-Zea mays, Sorghum bicolor and Setaria viridis. We show that single-cell and single-nucleus RNA sequencing provide complementary readouts of cell identity in dicots and monocots, warranting a combined analysis. Cell types were mapped across species to identify robust, orthologous marker genes. The comparative cellular analysis shows that the transcriptomes of some cell types diverged more rapidly than those of others-driven, in part, by recruitment of gene modules from other cell types. The data also show that a recent whole-genome duplication provides a rich source of new, highly localized gene expression domains that favour fast-evolving cell types. Together, the cell-by-cell comparative analysis shows how fine-scale cellular profiling can extract conserved modules from a pan transcriptome and provide insight on the evolution of cells that mediate key functions in crops.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
References
-
- Rich-Griffin C et al. Single-Cell Transcriptomics: A High-Resolution Avenue for Plant Functional Genomics. Trends Plant Sci. 25, 186–197 (2020). - PubMed
-
- Marioni JC & Arendt D How Single-Cell Genomics Is Changing Evolutionary and Developmental Biology. Annu. Rev. Cell Dev. Biol 33, 537–553 (2017). - PubMed
-
- Kajala K et al. Innovation, conservation, and repurposing of gene function in root cell type development. Cell 184, 3333–3348.e19 (2021). - PubMed
Methods References
-
- Hernández Coronado M et al. Repel or Repair: Plant Glutamate Receptor-Like Channels Mediate a Defense vs. Regeneration Tradeoff. SSRN Electron. J (2021). doi:10.2139/ssrn.3818443 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
