

Pan-cancer whole-genome comparison of primary and metastatic solid tumours
- PMID: 37165194
- PMCID: PMC10247378
- DOI: 10.1038/s41586-023-06054-z
Pan-cancer whole-genome comparison of primary and metastatic solid tumours
Abstract
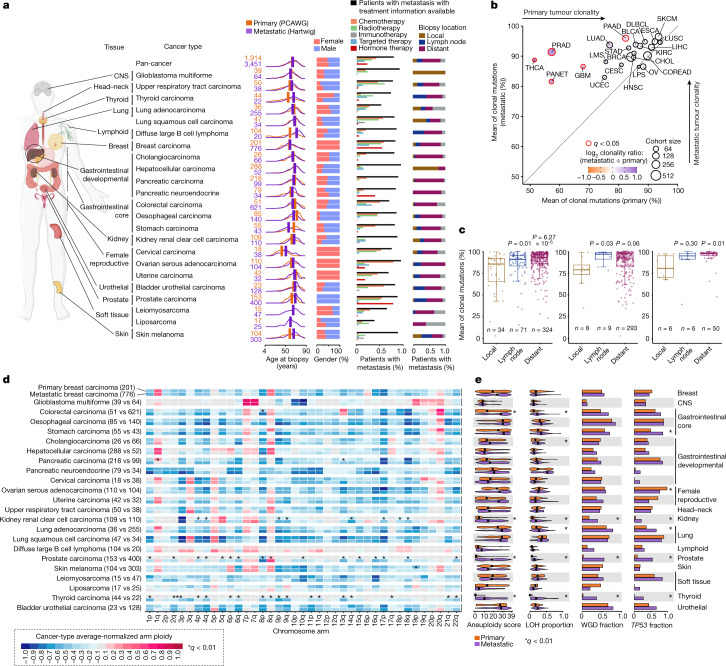
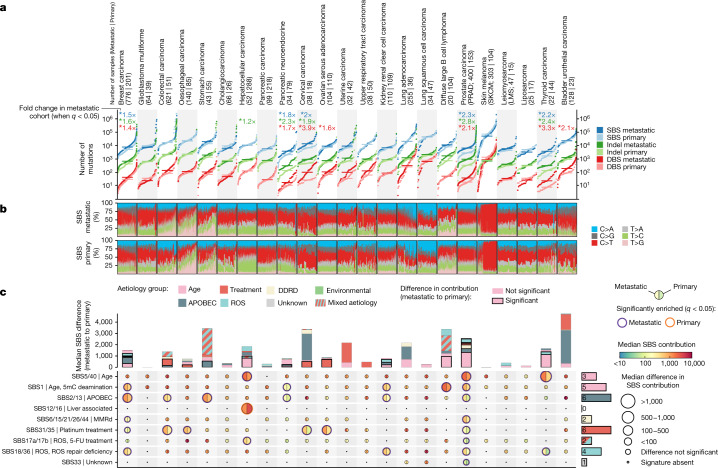
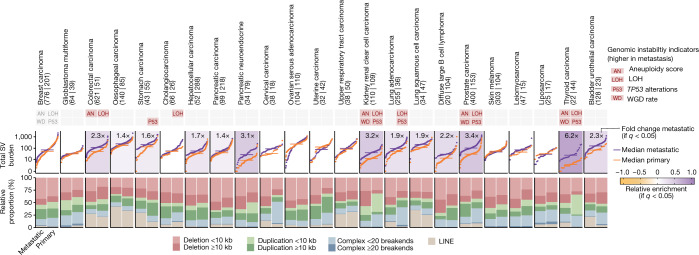
Metastatic cancer remains an almost inevitably lethal disease1-3. A better understanding of disease progression and response to therapies therefore remains of utmost importance. Here we characterize the genomic differences between early-stage untreated primary tumours and late-stage treated metastatic tumours using a harmonized pan-cancer analysis (or reanalysis) of two unpaired primary4 and metastatic5 cohorts of 7,108 whole-genome-sequenced tumours. Metastatic tumours in general have a lower intratumour heterogeneity and a conserved karyotype, displaying only a modest increase in mutations, although frequencies of structural variants are elevated overall. Furthermore, highly variable tumour-specific contributions of mutational footprints of endogenous (for example, SBS1 and APOBEC) and exogenous mutational processes (for example, platinum treatment) are present. The majority of cancer types had either moderate genomic differences (for example, lung adenocarcinoma) or highly consistent genomic portraits (for example, ovarian serous carcinoma) when comparing early-stage and late-stage disease. Breast, prostate, thyroid and kidney renal clear cell carcinomas and pancreatic neuroendocrine tumours are clear exceptions to the rule, displaying an extensive transformation of their genomic landscape in advanced stages. Exposure to treatment further scars the tumour genome and introduces an evolutionary bottleneck that selects for known therapy-resistant drivers in approximately half of treated patients. Our data showcase the potential of pan-cancer whole-genome analysis to identify distinctive features of late-stage tumours and provide a valuable resource to further investigate the biological basis of cancer and resistance to therapies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












Comment in
-
Primary, Metastatic Tumors Vary Genomically by Cancer Type.Cancer Discov. 2023 Jul 7;13(7):1505-1506. doi: 10.1158/2159-8290.CD-NB2023-0041. Cancer Discov. 2023. PMID: 37249311
-
The evolving cancer genome.Nat Genet. 2023 Jul;55(7):1082. doi: 10.1038/s41588-023-01457-0. Nat Genet. 2023. PMID: 37438537 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
