

Widespread somatic L1 retrotransposition in normal colorectal epithelium
- PMID: 37165195
- PMCID: PMC10191854
- DOI: 10.1038/s41586-023-06046-z
Widespread somatic L1 retrotransposition in normal colorectal epithelium
Abstract
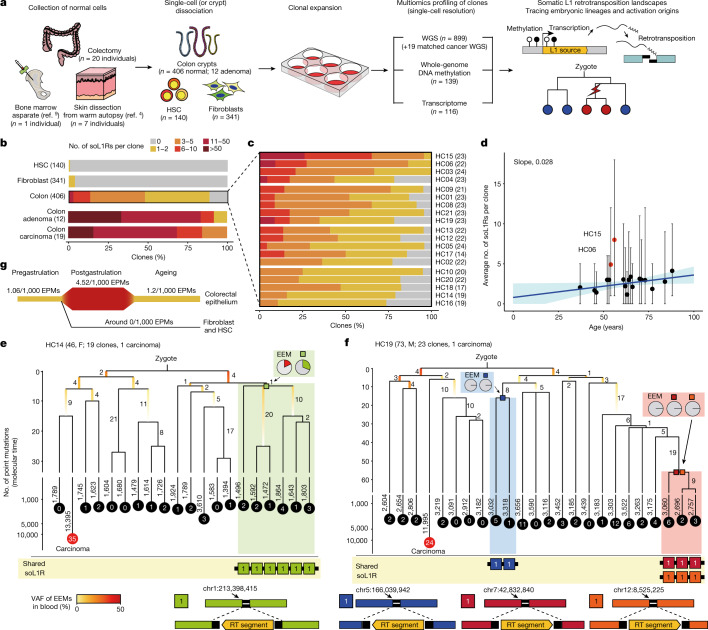
Throughout an individual's lifetime, genomic alterations accumulate in somatic cells1-11. However, the mutational landscape induced by retrotransposition of long interspersed nuclear element-1 (L1), a widespread mobile element in the human genome12-14, is poorly understood in normal cells. Here we explored the whole-genome sequences of 899 single-cell clones established from three different cell types collected from 28 individuals. We identified 1,708 somatic L1 retrotransposition events that were enriched in colorectal epithelium and showed a positive relationship with age. Fingerprinting of source elements showed 34 retrotransposition-competent L1s. Multidimensional analysis demonstrated that (1) somatic L1 retrotranspositions occur from early embryogenesis at a substantial rate, (2) epigenetic on/off of a source element is preferentially determined in the early organogenesis stage, (3) retrotransposition-competent L1s with a lower population allele frequency have higher retrotransposition activity and (4) only a small fraction of L1 transcripts in the cytoplasm are finally retrotransposed in somatic cells. Analysis of matched cancers further suggested that somatic L1 retrotransposition rate is substantially increased during colorectal tumourigenesis. In summary, this study illustrates L1 retrotransposition-induced somatic mosaicism in normal cells and provides insights into the genomic and epigenomic regulation of transposable elements over the human lifetime.
© 2023. The Author(s).
Conflict of interest statement
Y.S.J. is a cofounder and chief executive officer of Genome Insight, Inc. The remaining authors declare no competing interests.
Figures















References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
