

Increased mutation and gene conversion within human segmental duplications
- PMID: 37165237
- PMCID: PMC10172114
- DOI: 10.1038/s41586-023-05895-y
Increased mutation and gene conversion within human segmental duplications
Abstract
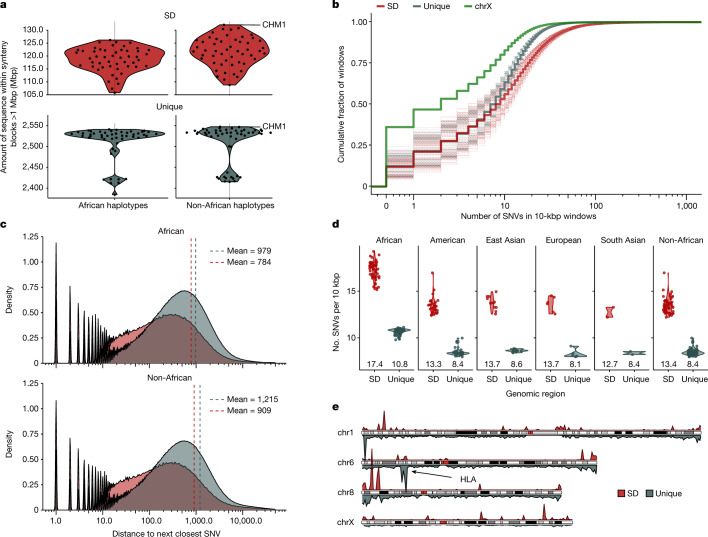
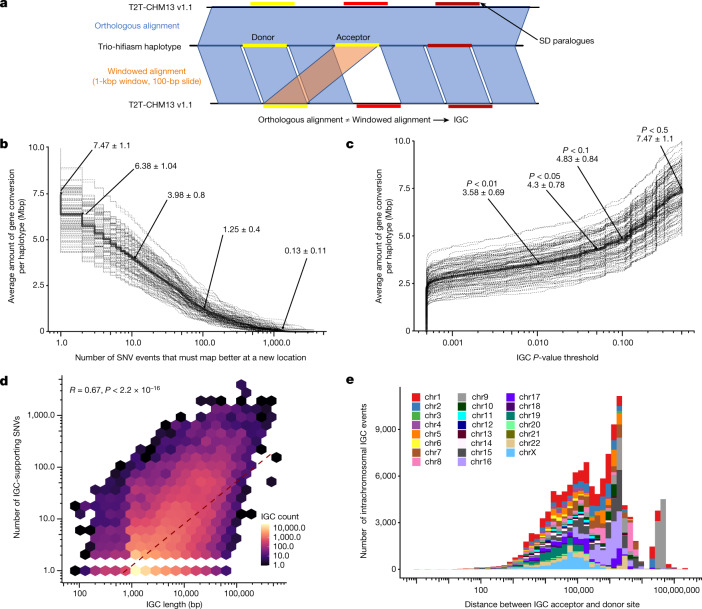
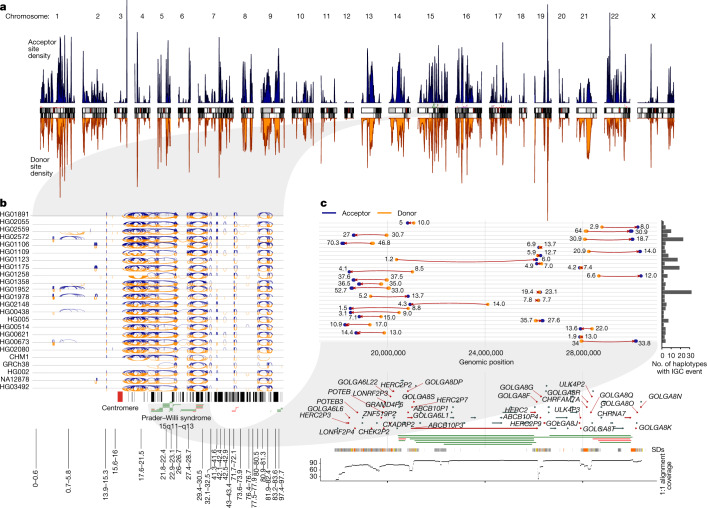
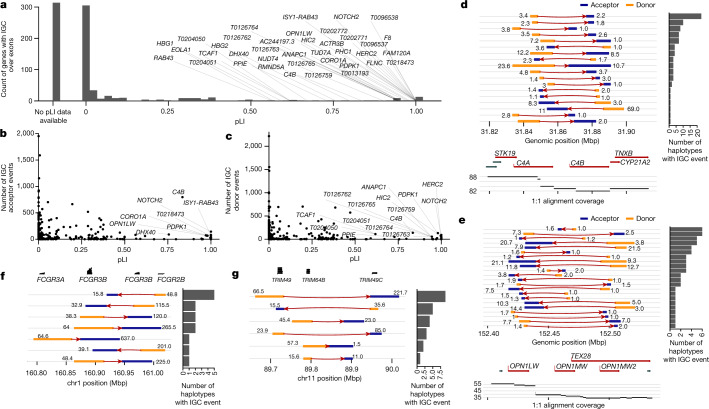
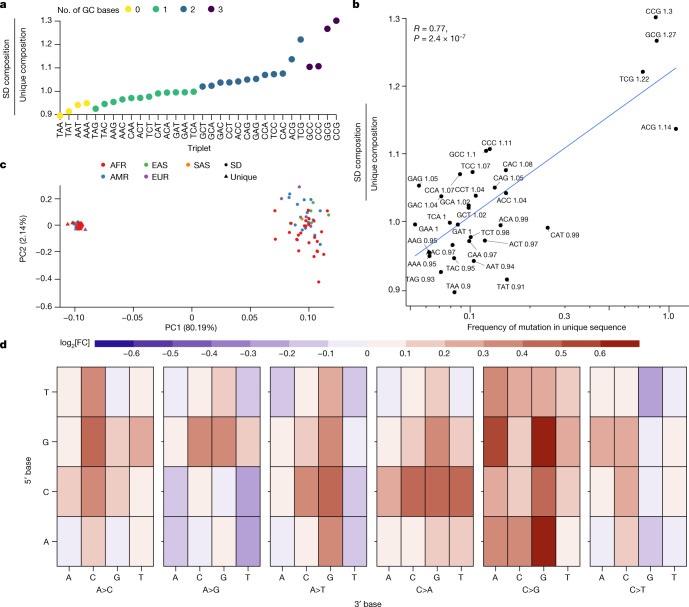
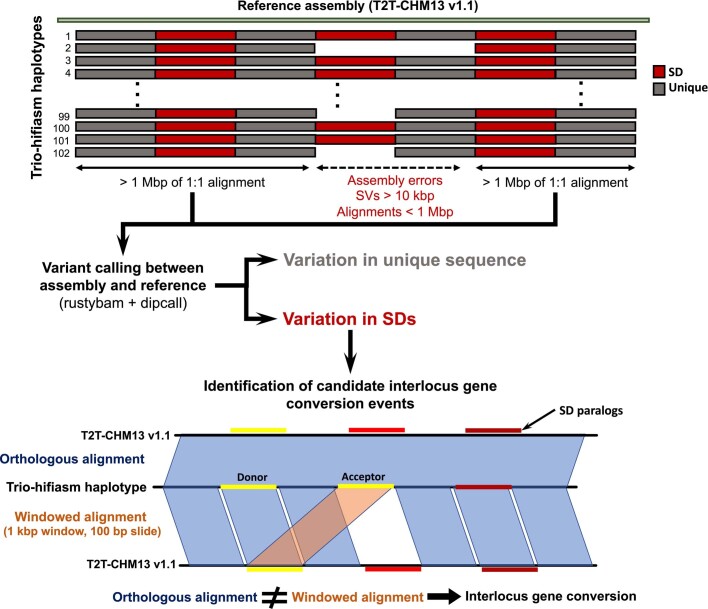
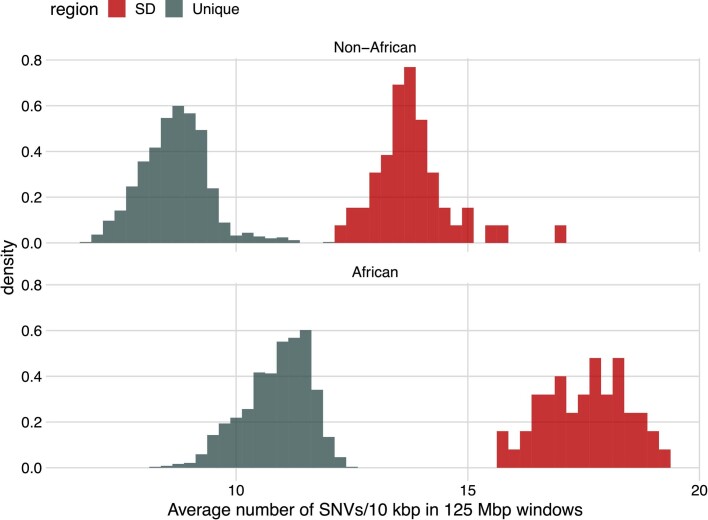
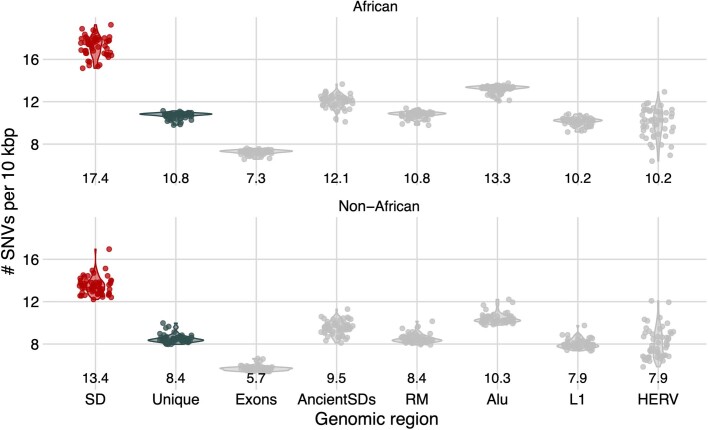
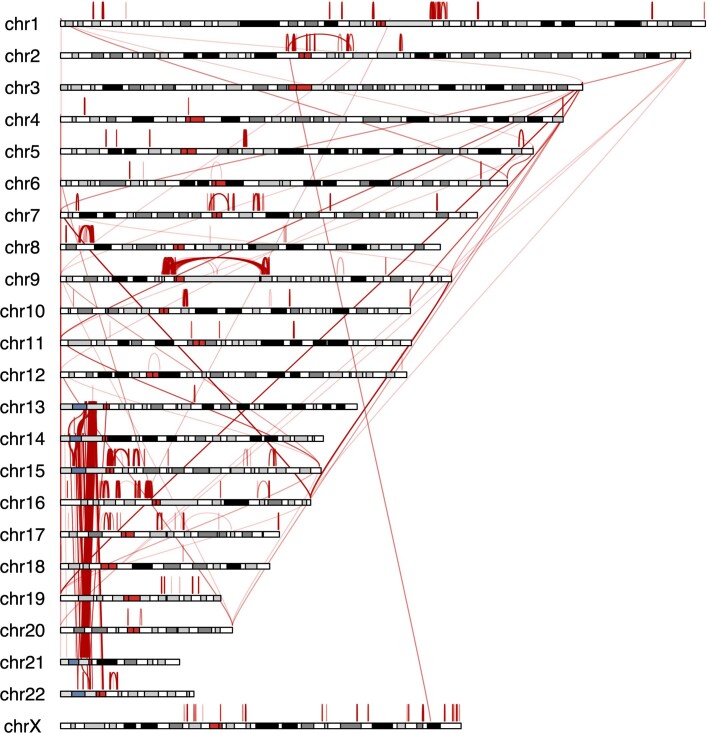
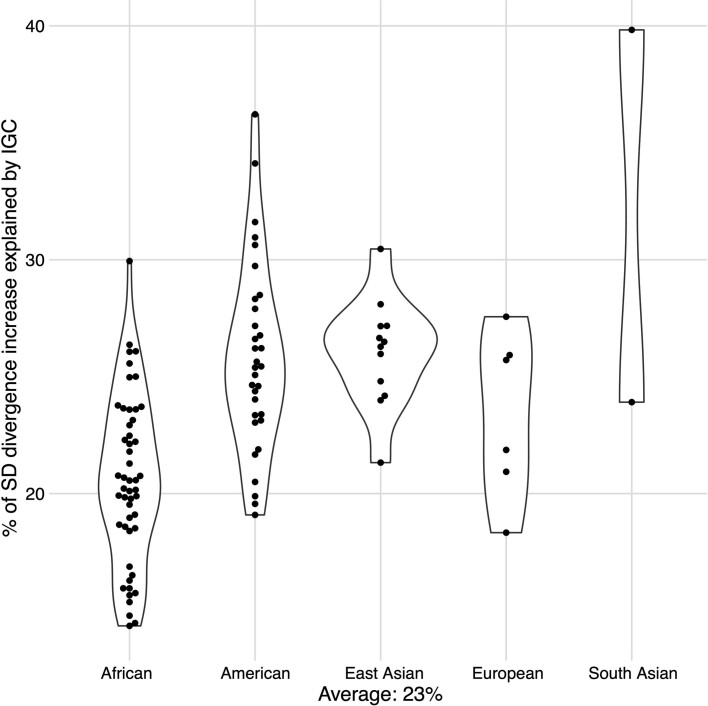
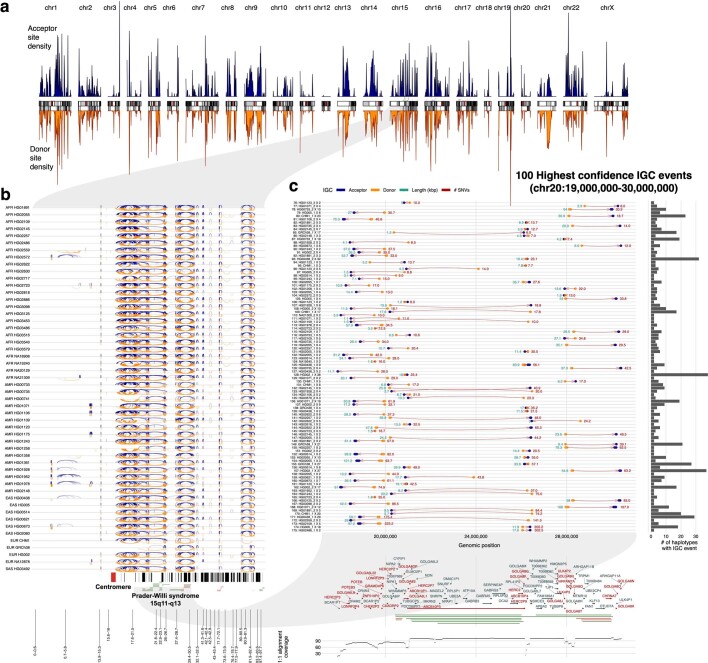
Single-nucleotide variants (SNVs) in segmental duplications (SDs) have not been systematically assessed because of the limitations of mapping short-read sequencing data1,2. Here we constructed 1:1 unambiguous alignments spanning high-identity SDs across 102 human haplotypes and compared the pattern of SNVs between unique and duplicated regions3,4. We find that human SNVs are elevated 60% in SDs compared to unique regions and estimate that at least 23% of this increase is due to interlocus gene conversion (IGC) with up to 4.3 megabase pairs of SD sequence converted on average per human haplotype. We develop a genome-wide map of IGC donors and acceptors, including 498 acceptor and 454 donor hotspots affecting the exons of about 800 protein-coding genes. These include 171 genes that have 'relocated' on average 1.61 megabase pairs in a subset of human haplotypes. Using a coalescent framework, we show that SD regions are slightly evolutionarily older when compared to unique sequences, probably owing to IGC. SNVs in SDs, however, show a distinct mutational spectrum: a 27.1% increase in transversions that convert cytosine to guanine or the reverse across all triplet contexts and a 7.6% reduction in the frequency of CpG-associated mutations when compared to unique DNA. We reason that these distinct mutational properties help to maintain an overall higher GC content of SD DNA compared to that of unique DNA, probably driven by GC-biased conversion between paralogous sequences5,6.
© 2023. The Author(s).
Conflict of interest statement
E.E.E. is a scientific advisory board member of Variant Bio, Inc. All other authors declare no competing interests.
Figures

Comment in
-
Human pangenome supports analysis of complex genomic regions.Nature. 2023 May;617(7960):256-258. doi: 10.1038/d41586-023-01490-3. Nature. 2023. PMID: 37165235 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
