

A draft human pangenome reference
- PMID: 37165242
- PMCID: PMC10172123
- DOI: 10.1038/s41586-023-05896-x
A draft human pangenome reference
Abstract
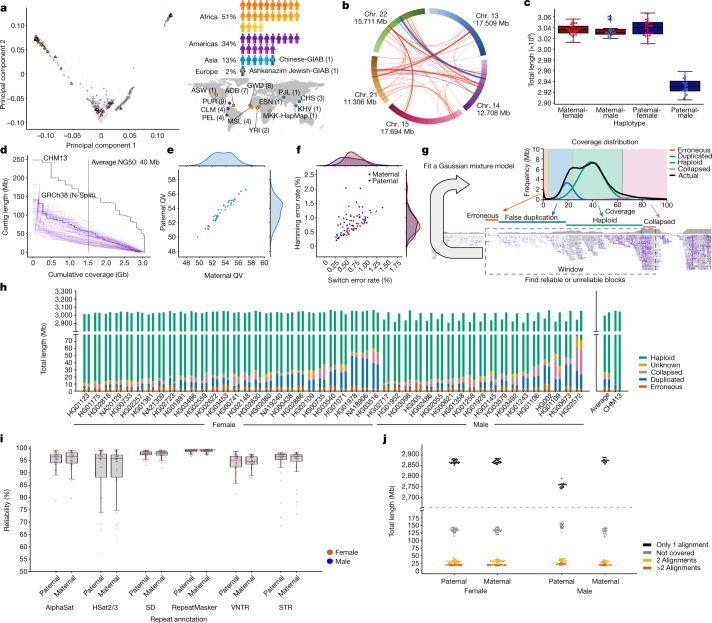
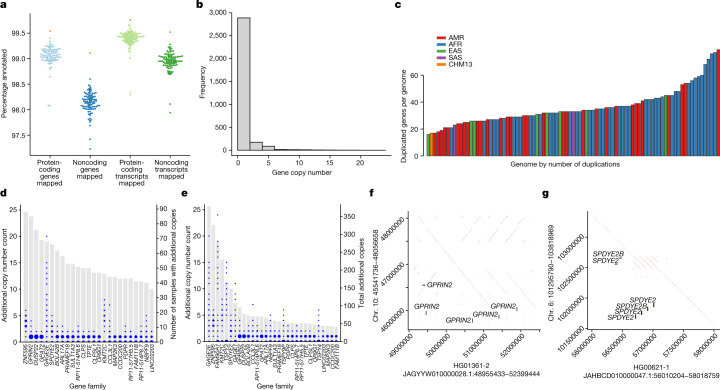
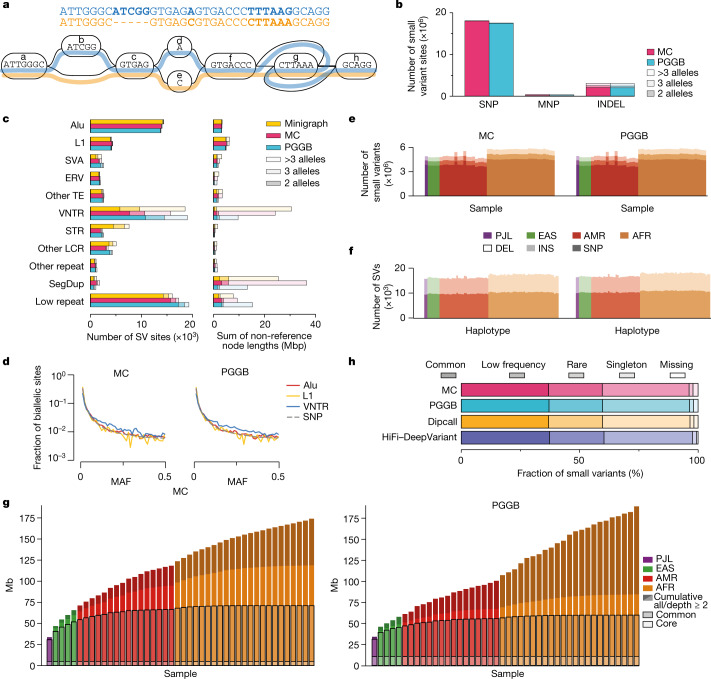
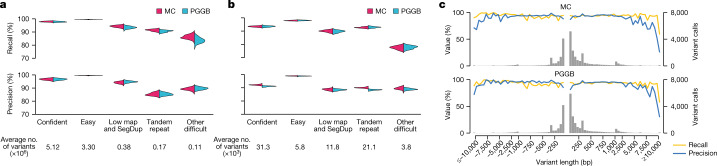
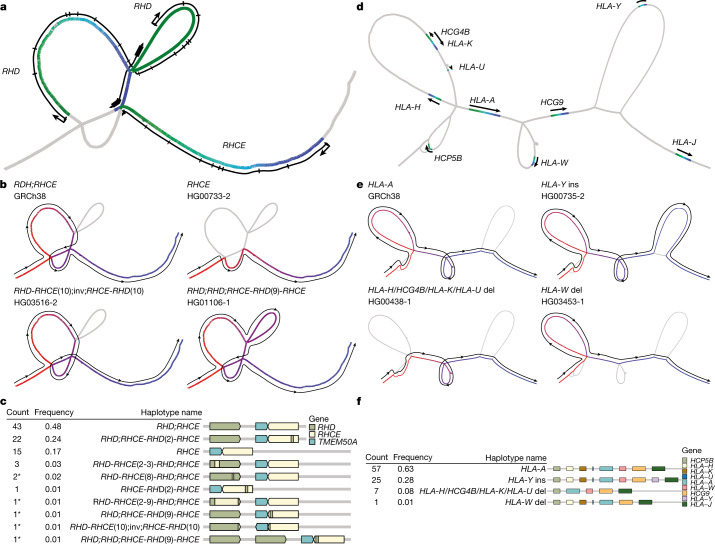
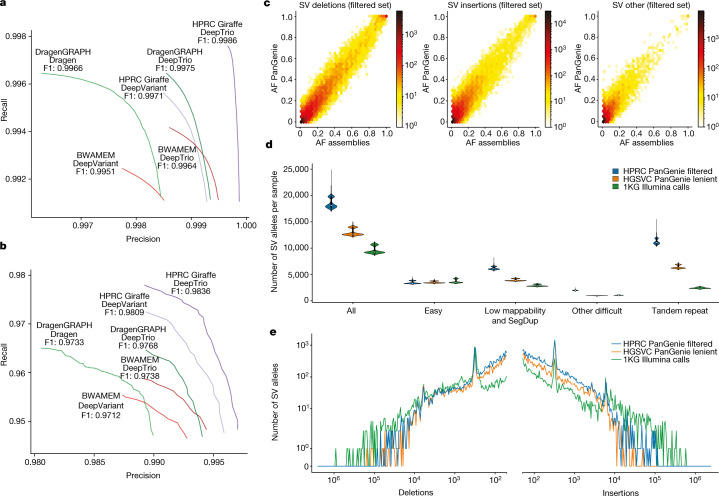
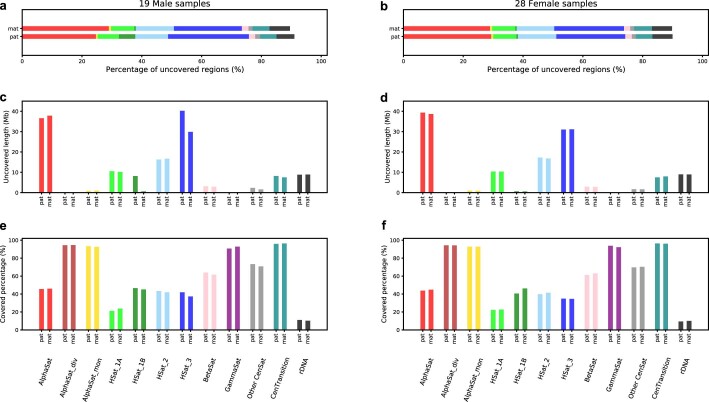
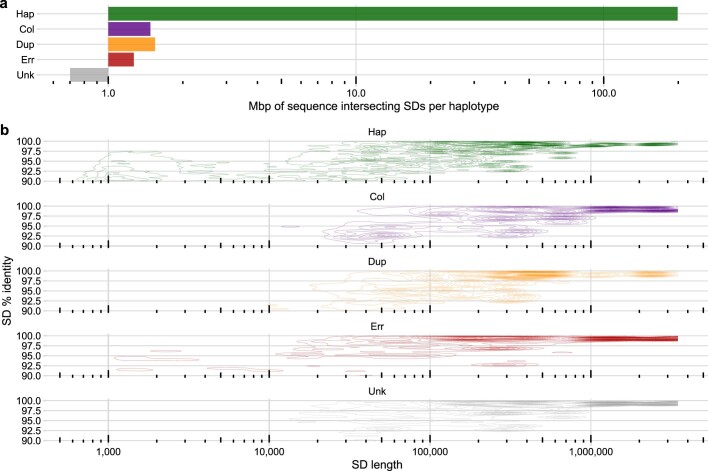
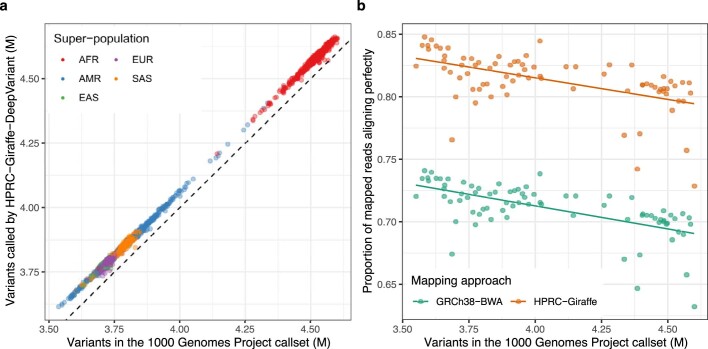
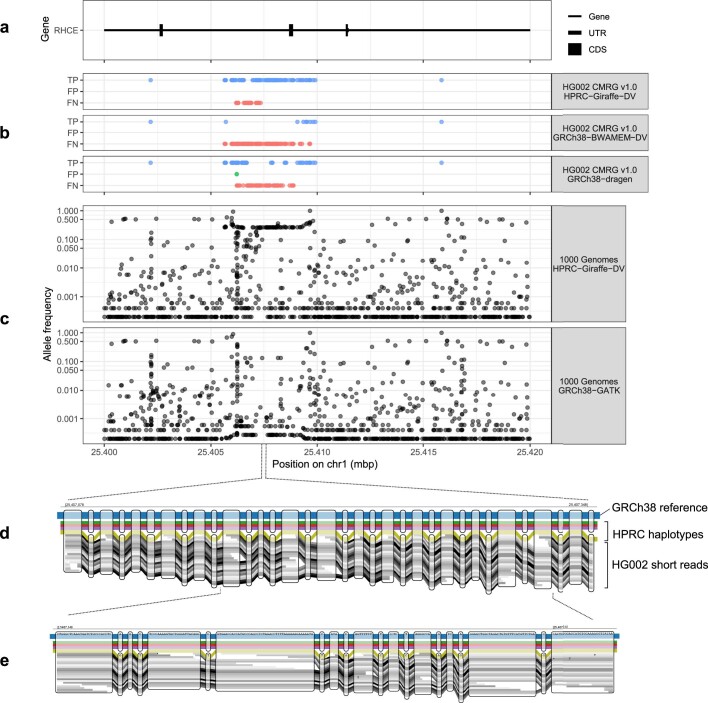
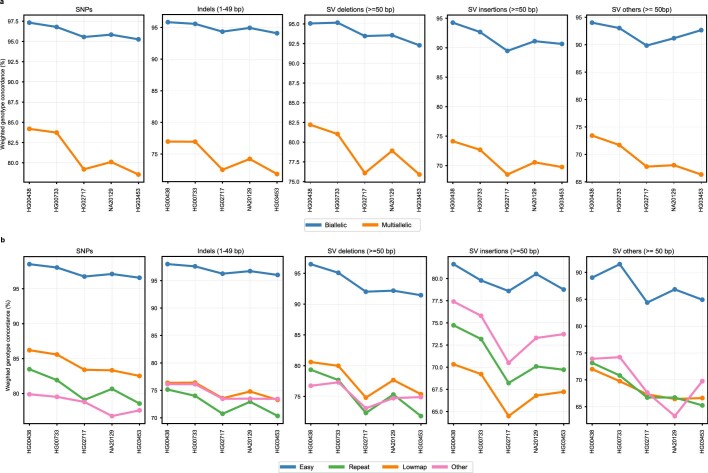
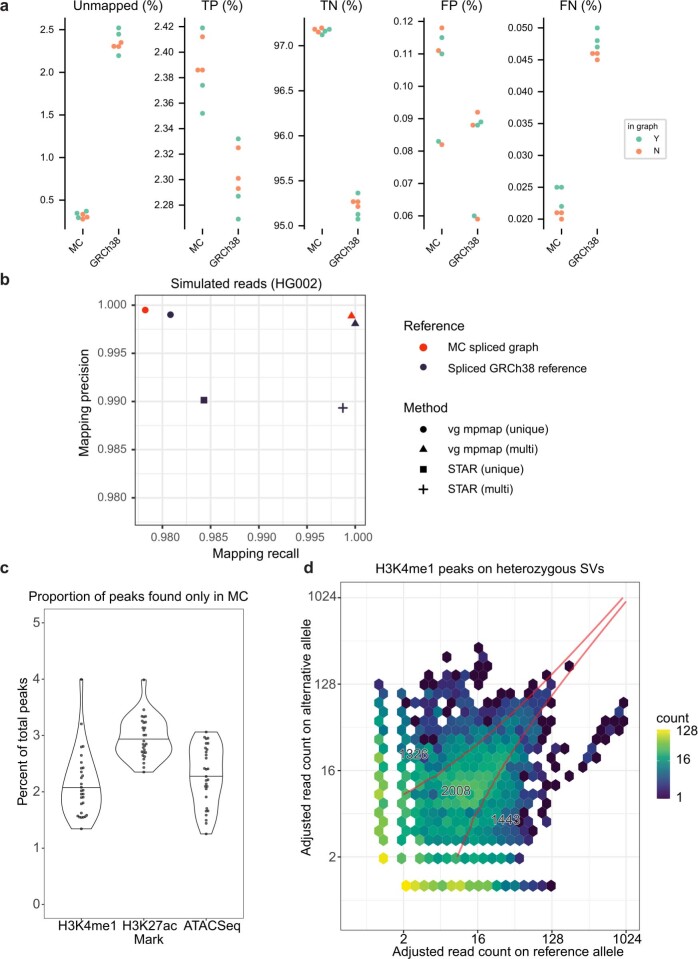
Here the Human Pangenome Reference Consortium presents a first draft of the human pangenome reference. The pangenome contains 47 phased, diploid assemblies from a cohort of genetically diverse individuals1. These assemblies cover more than 99% of the expected sequence in each genome and are more than 99% accurate at the structural and base pair levels. Based on alignments of the assemblies, we generate a draft pangenome that captures known variants and haplotypes and reveals new alleles at structurally complex loci. We also add 119 million base pairs of euchromatic polymorphic sequences and 1,115 gene duplications relative to the existing reference GRCh38. Roughly 90 million of the additional base pairs are derived from structural variation. Using our draft pangenome to analyse short-read data reduced small variant discovery errors by 34% and increased the number of structural variants detected per haplotype by 104% compared with GRCh38-based workflows, which enabled the typing of the vast majority of structural variant alleles per sample.
© 2023. The Author(s).
Conflict of interest statement
E.E.E. is a scientific advisory board (SAB) member of Variant Bio. P.F is a member of the SABs of Fabric Genomics and Eagle Genomics. E.E.K. is a member of the SAB of Encompass Biosciences, Foresite Labs and Galateo Bio and has received personal fees from Regeneron Pharmaceuticals, 23&Me and Illumina. A.B., A.C., P.-C.C., D.E.C., G.Baid, A.K., M.N. and K.S. are employees of Google and own Alphabet stock as part of the standard compensation package.
Figures
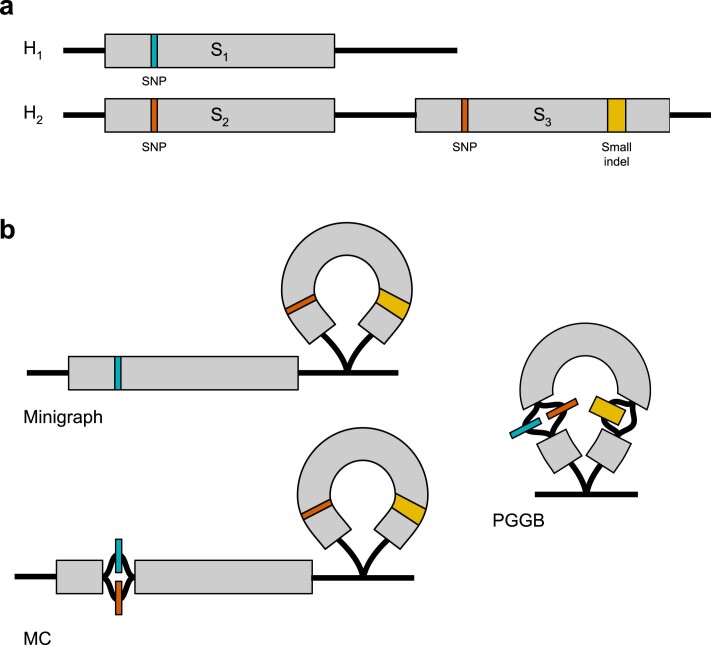
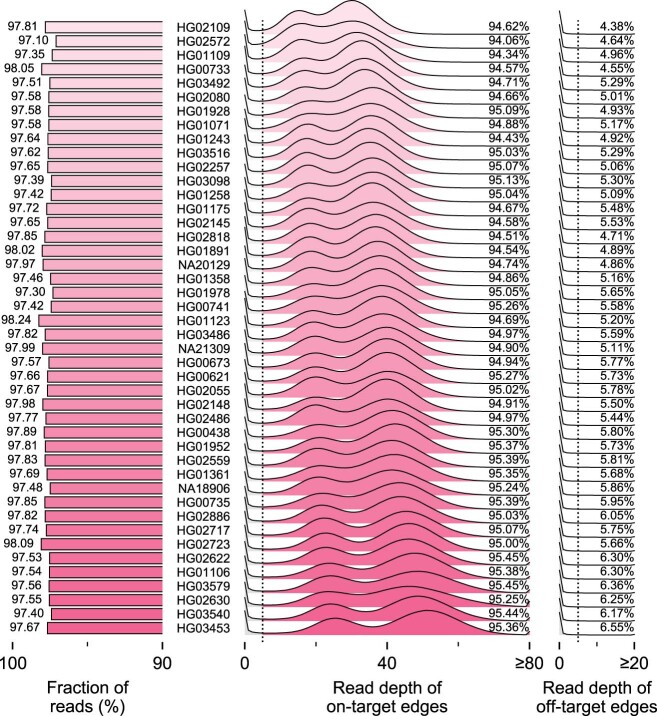
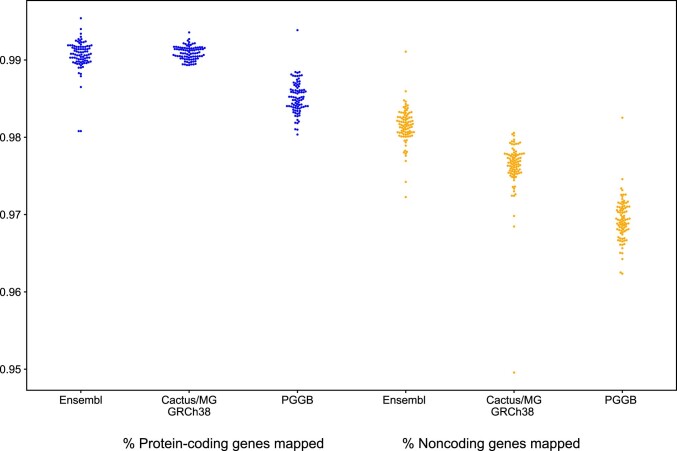
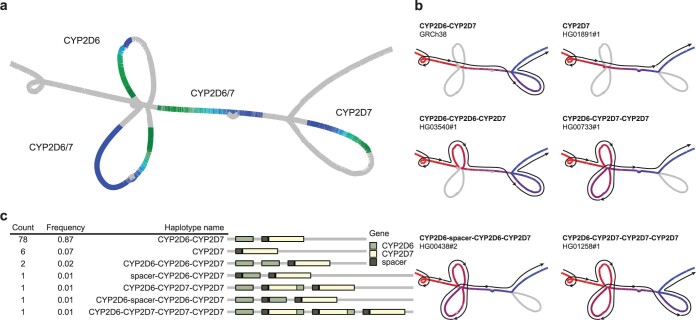
Comment in
-
Human pangenome supports analysis of complex genomic regions.Nature. 2023 May;617(7960):256-258. doi: 10.1038/d41586-023-01490-3. Nature. 2023. PMID: 37165235 No abstract available.
-
New Genomic Sequencing Resource Could Improve Care.Cancer Discov. 2023 Jul 7;13(7):1506-1507. doi: 10.1158/2159-8290.CD-NB2023-0042. Cancer Discov. 2023. PMID: 37249320
References
Publication types
MeSH terms
Grants and funding
- R01 HG006677/HG/NHGRI NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- R35 GM130151/GM/NIGMS NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- U01 HG010973/HG/NHGRI NIH HHS/United States
- R01 HG011649/HG/NHGRI NIH HHS/United States
- U01 HG010963/HG/NHGRI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U24 HG009081/HG/NHGRI NIH HHS/United States
- U24 HG010262/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- U24 HG011853/HG/NHGRI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U24 HG007497/HG/NHGRI NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- ZIA HG200398/ImNIH/Intramural NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- OT2 OD033761/OD/NIH HHS/United States
- R01 GM123489/GM/NIGMS NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
