

Gluconeogenic enzyme PCK1 supports S-adenosylmethionine biosynthesis and promotes H3K9me3 modification to suppress hepatocellular carcinoma progression
- PMID: 37166978
- PMCID: PMC10313362
- DOI: 10.1172/JCI161713
Gluconeogenic enzyme PCK1 supports S-adenosylmethionine biosynthesis and promotes H3K9me3 modification to suppress hepatocellular carcinoma progression
Abstract
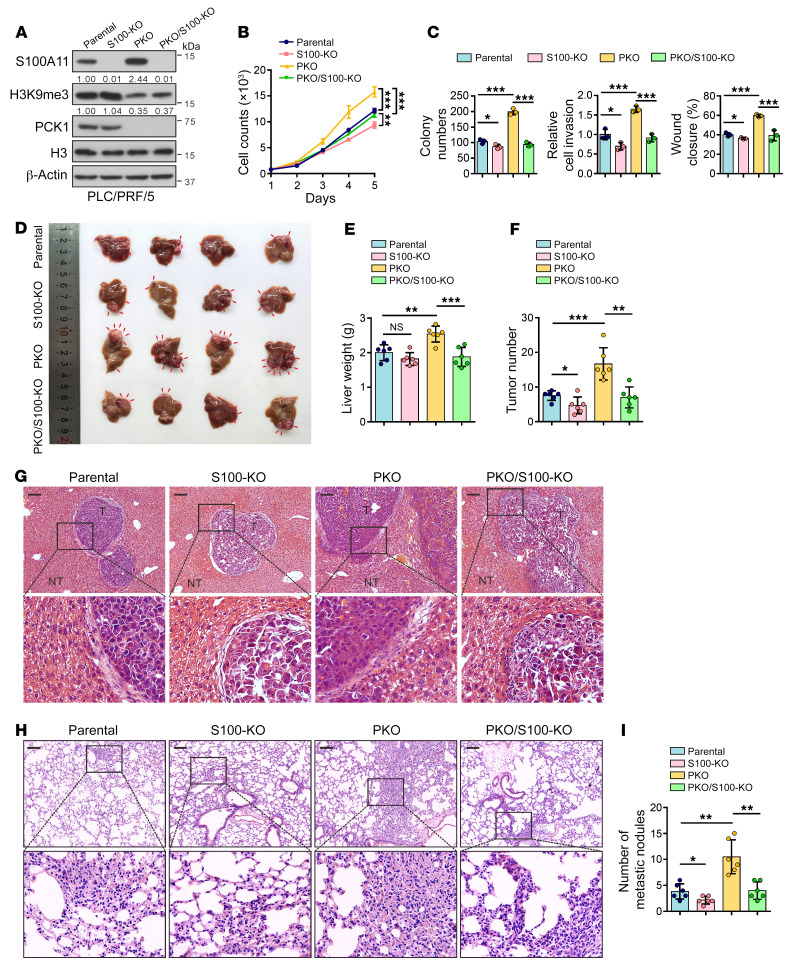
Deciphering the crosstalk between metabolic reprogramming and epigenetic regulation is a promising strategy for cancer therapy. In this study, we discovered that the gluconeogenic enzyme PCK1 fueled the generation of S-adenosylmethionine (SAM) through the serine synthesis pathway. The methyltransferase SUV39H1 catalyzed SAM, which served as a methyl donor to support H3K9me3 modification, leading to the suppression of the oncogene S100A11. Mechanistically, PCK1 deficiency-induced oncogenic activation of S100A11 was due to its interaction with AKT1, which upregulated PI3K/AKT signaling. Intriguingly, the progression of hepatocellular carcinoma (HCC) driven by PCK1 deficiency was suppressed by SAM supplement or S100A11 KO in vivo and in vitro. These findings reveal the availability of the key metabolite SAM as a bridge connecting the gluconeogenic enzyme PCK1 and H3K9 trimethylation in attenuating HCC progression, thus suggesting a potential therapeutic strategy against HCC.
Keywords: Gluconeogenesis; Liver cancer; Metabolism; Oncology.
Figures








References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
