

Integration of a multi-omics stem cell differentiation dataset using a dynamical model
- PMID: 37167320
- PMCID: PMC10204997
- DOI: 10.1371/journal.pgen.1010744
Integration of a multi-omics stem cell differentiation dataset using a dynamical model
Abstract
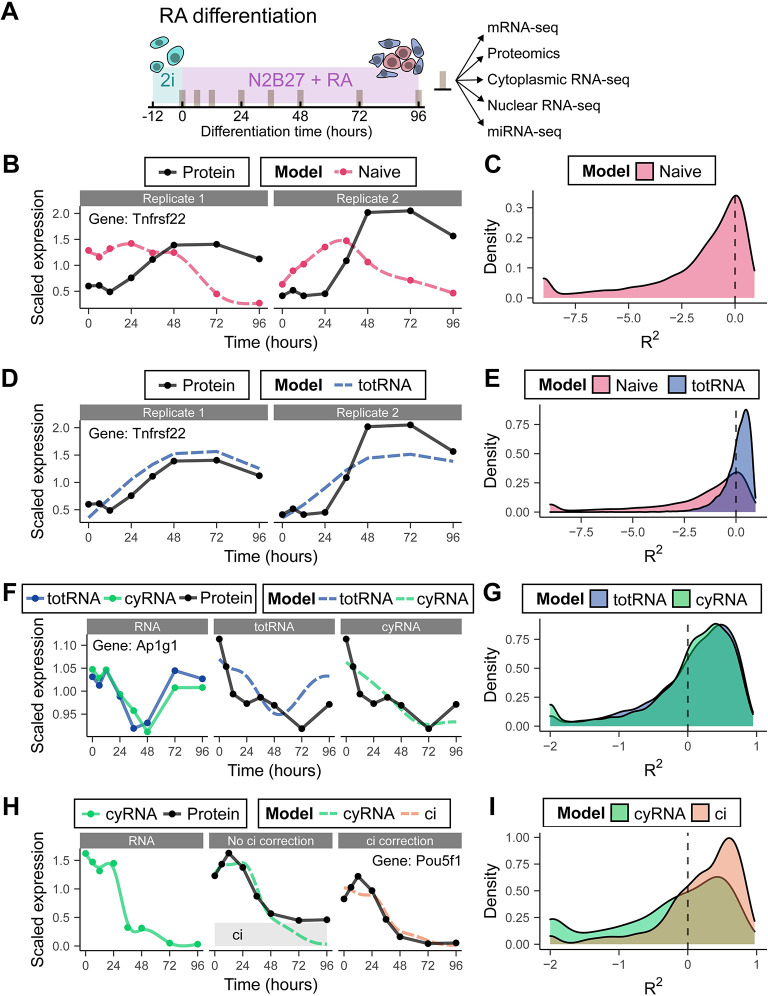
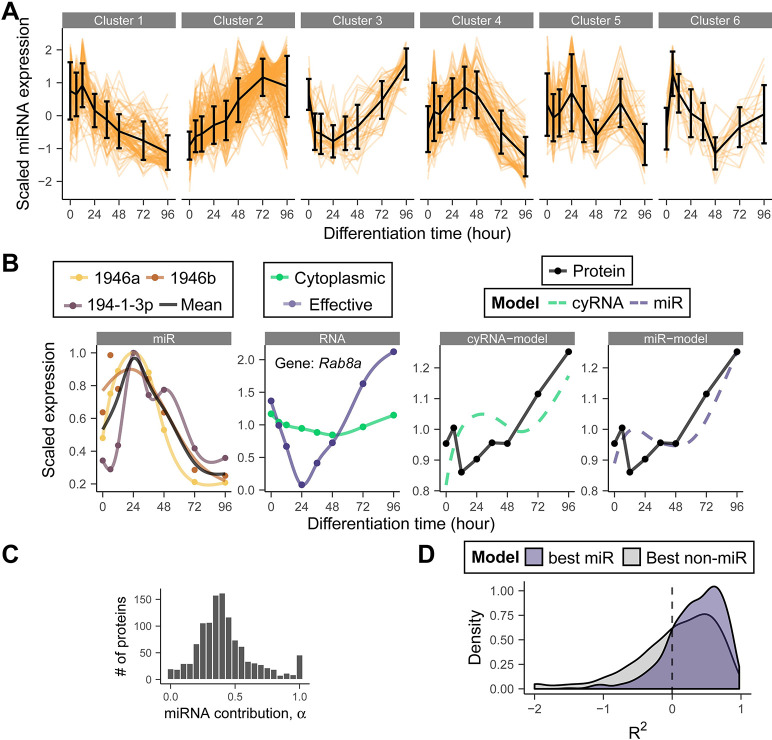
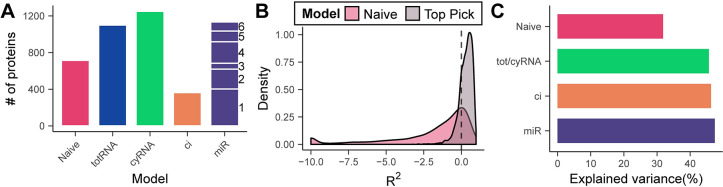
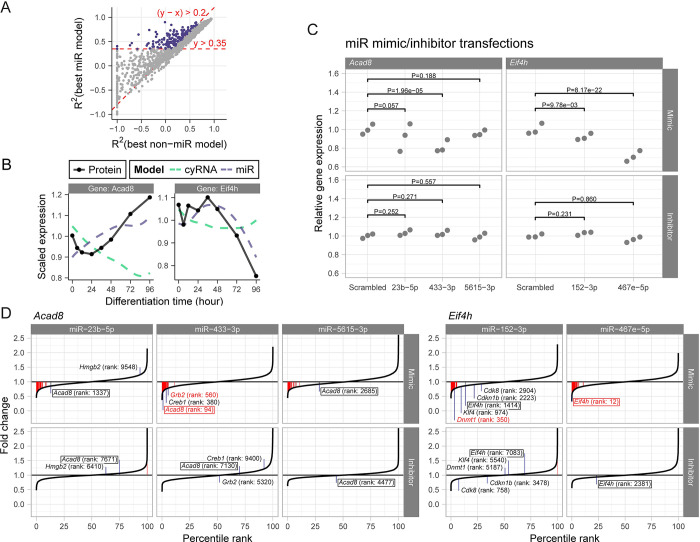
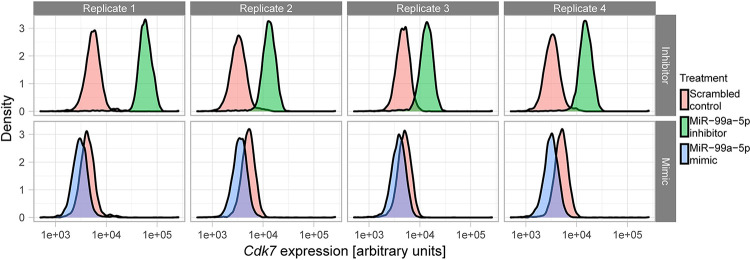
Stem cell differentiation is a highly dynamic process involving pervasive changes in gene expression. The large majority of existing studies has characterized differentiation at the level of individual molecular profiles, such as the transcriptome or the proteome. To obtain a more comprehensive view, we measured protein, mRNA and microRNA abundance during retinoic acid-driven differentiation of mouse embryonic stem cells. We found that mRNA and protein abundance are typically only weakly correlated across time. To understand this finding, we developed a hierarchical dynamical model that allowed us to integrate all data sets. This model was able to explain mRNA-protein discordance for most genes and identified instances of potential microRNA-mediated regulation. Overexpression or depletion of microRNAs identified by the model, followed by RNA sequencing and protein quantification, were used to follow up on the predictions of the model. Overall, our study shows how multi-omics integration by a dynamical model could be used to nominate candidate regulators.
Copyright: © 2023 van den Berg et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
