

Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis
- PMID: 37167403
- PMCID: PMC10316746
- DOI: 10.1126/science.add5327
Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis
Abstract
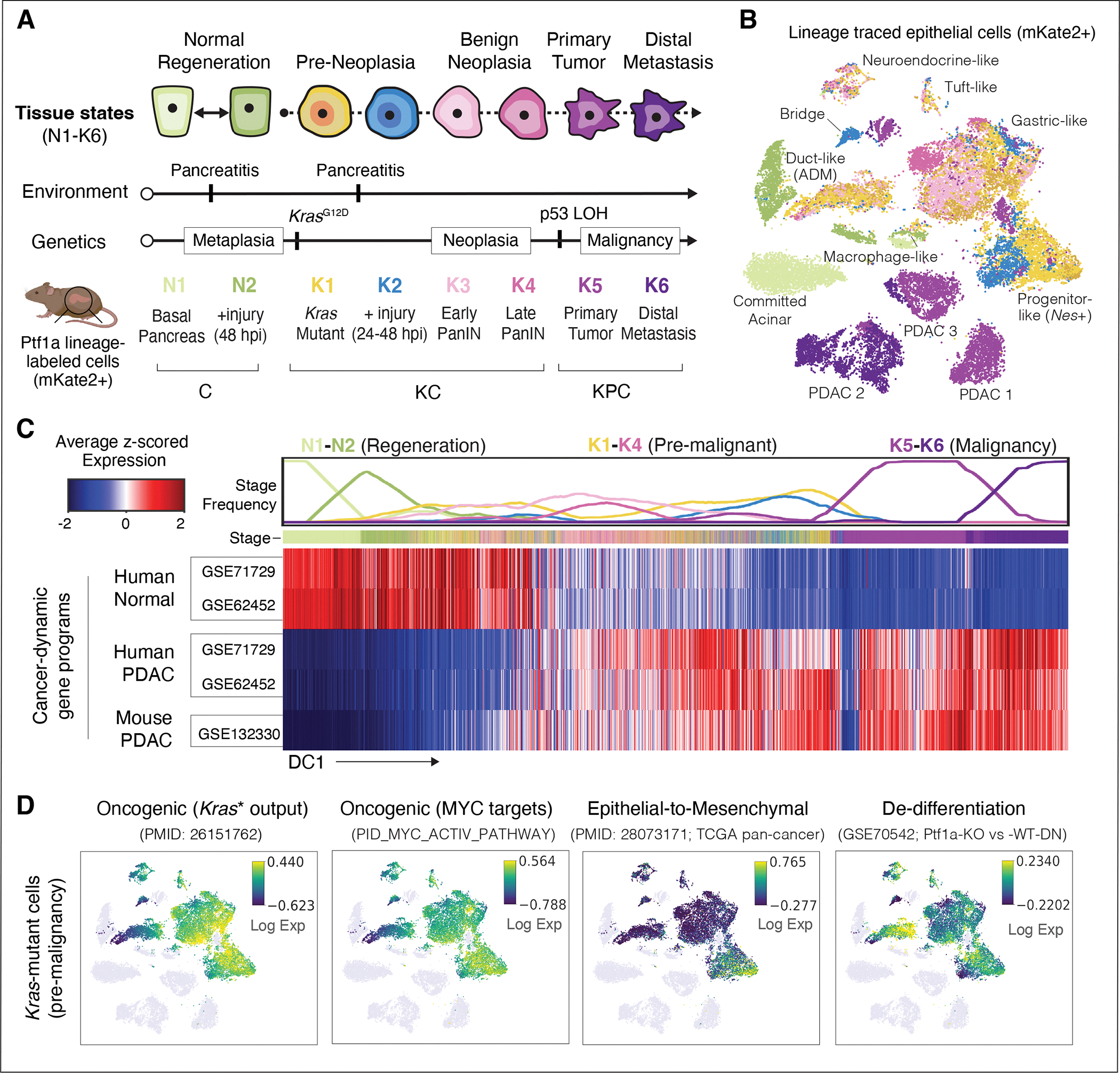
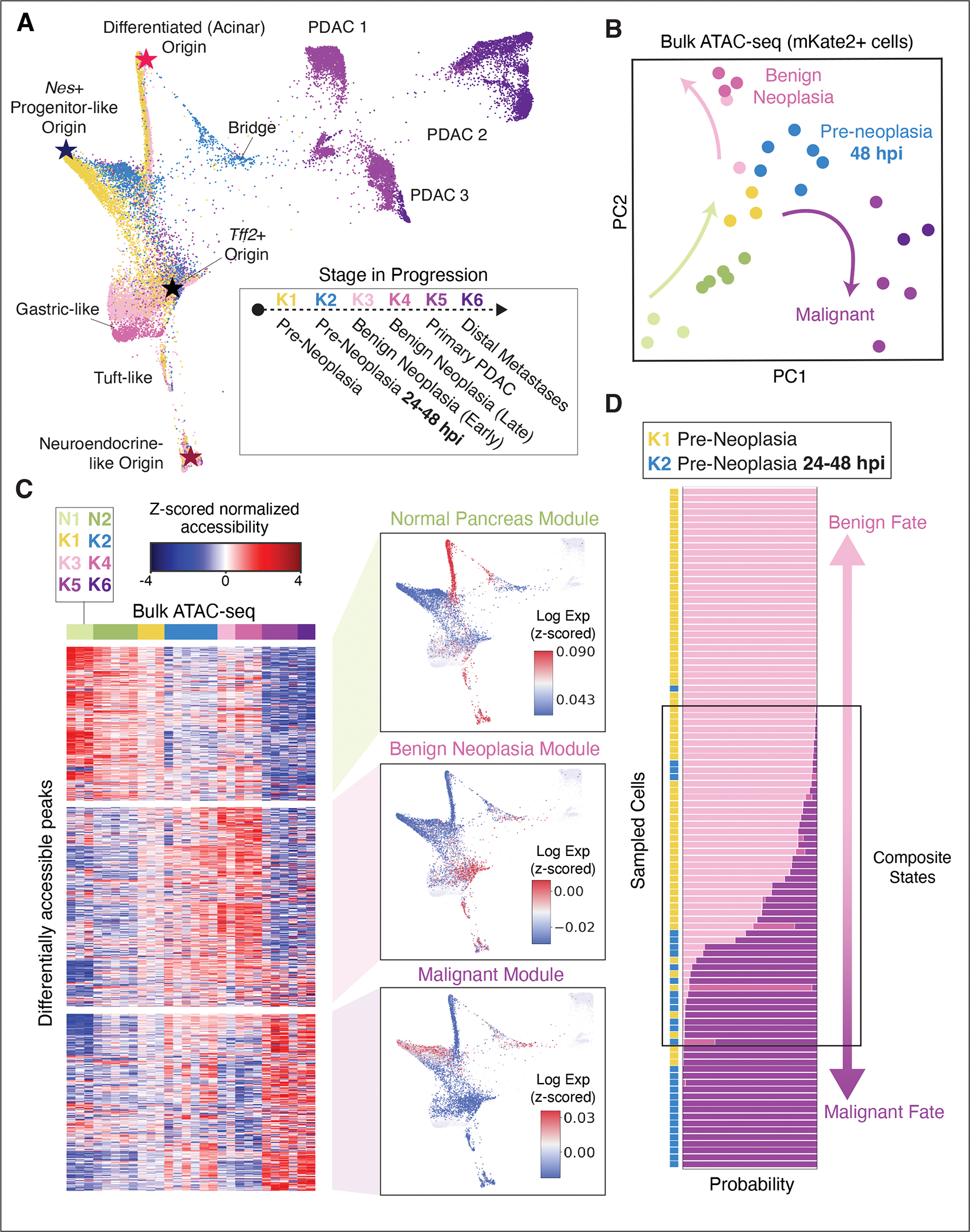
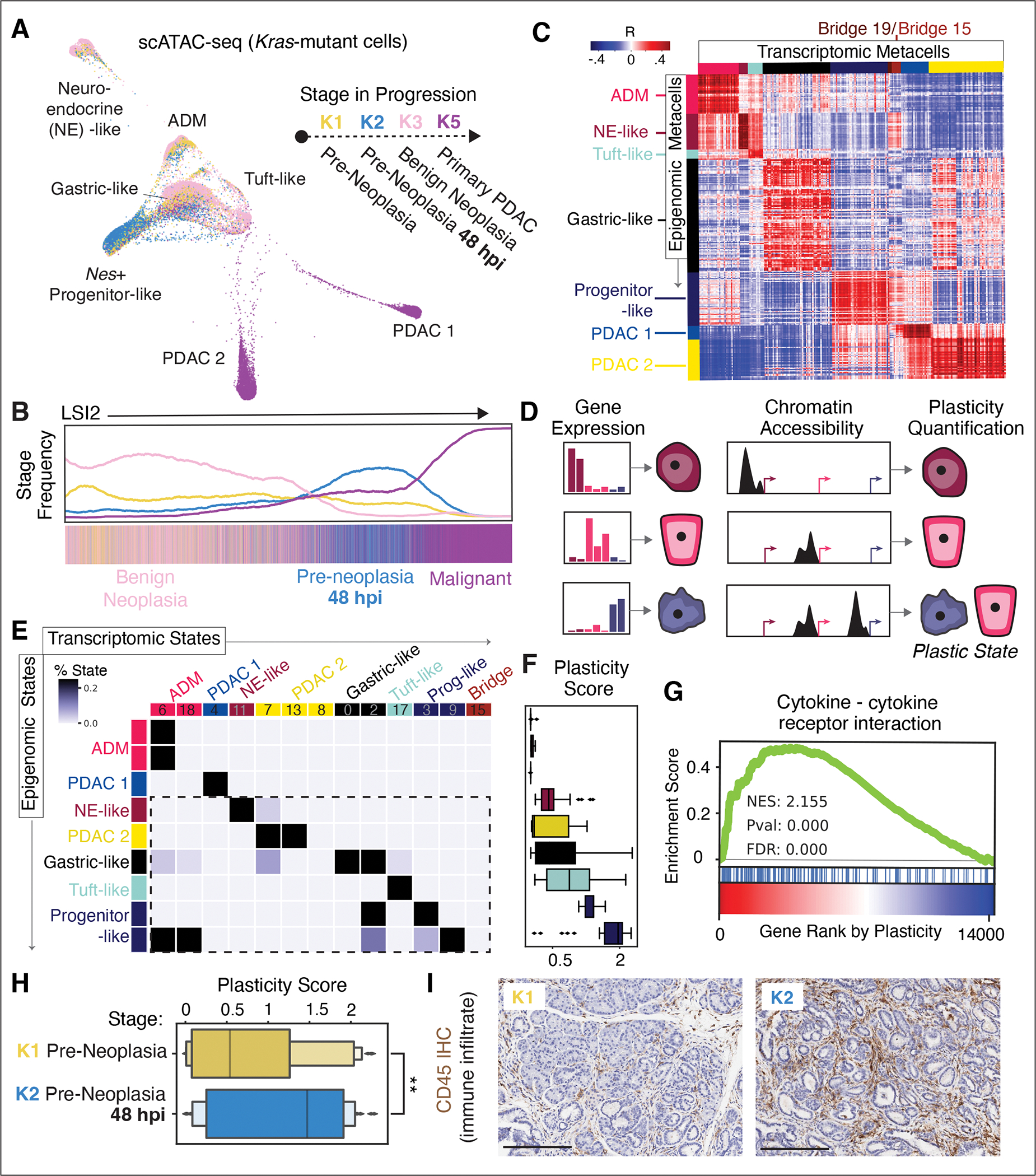
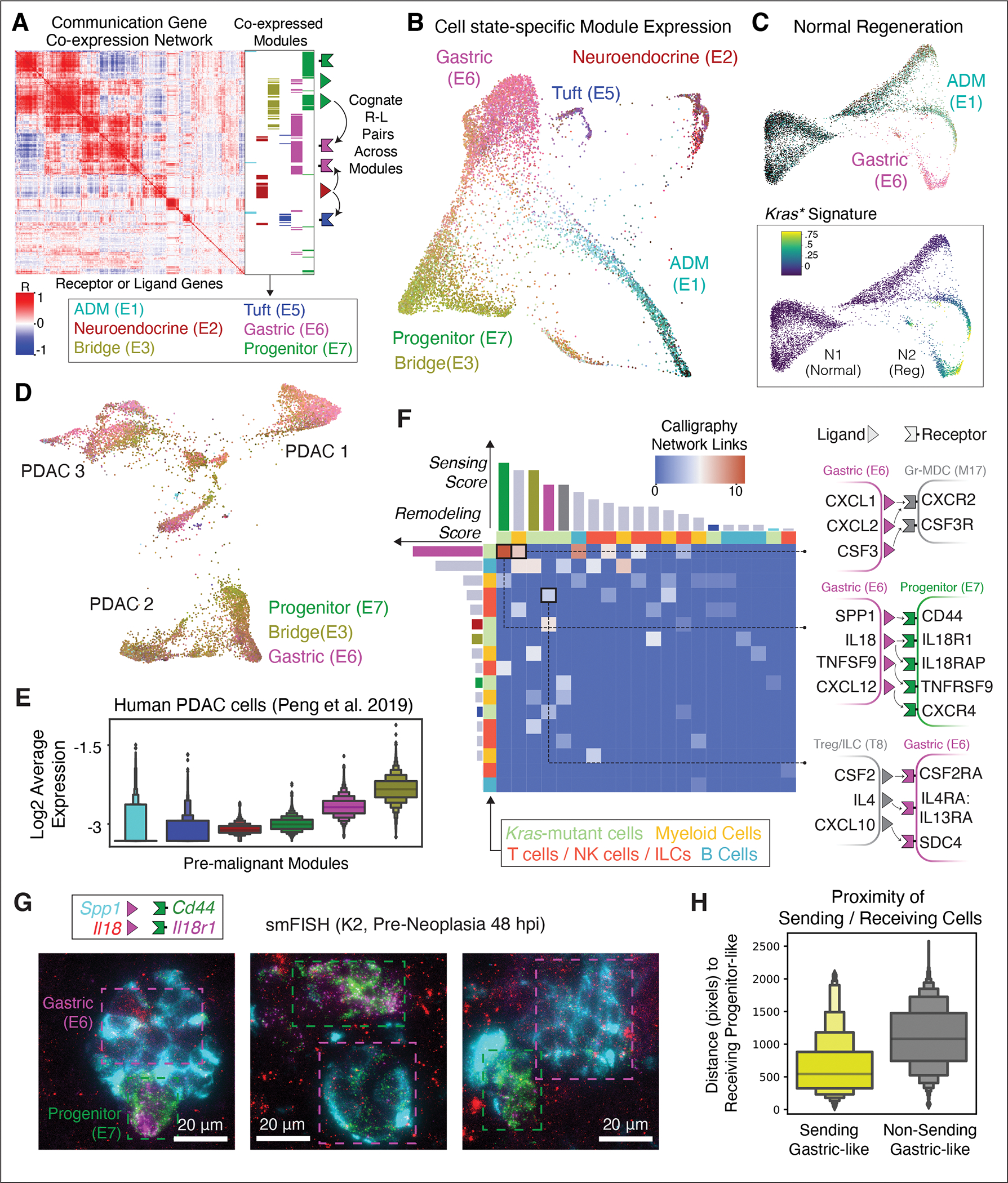
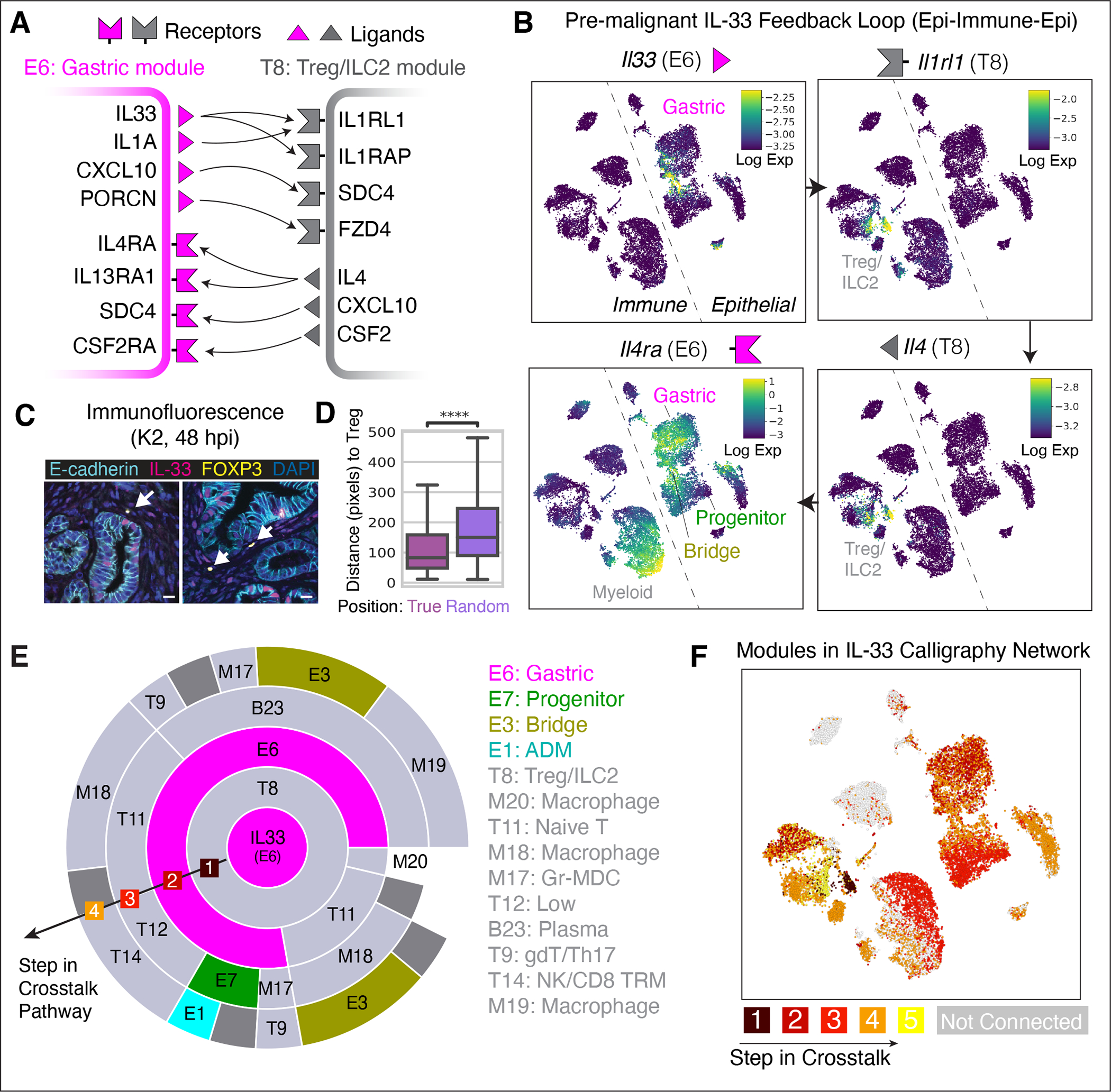
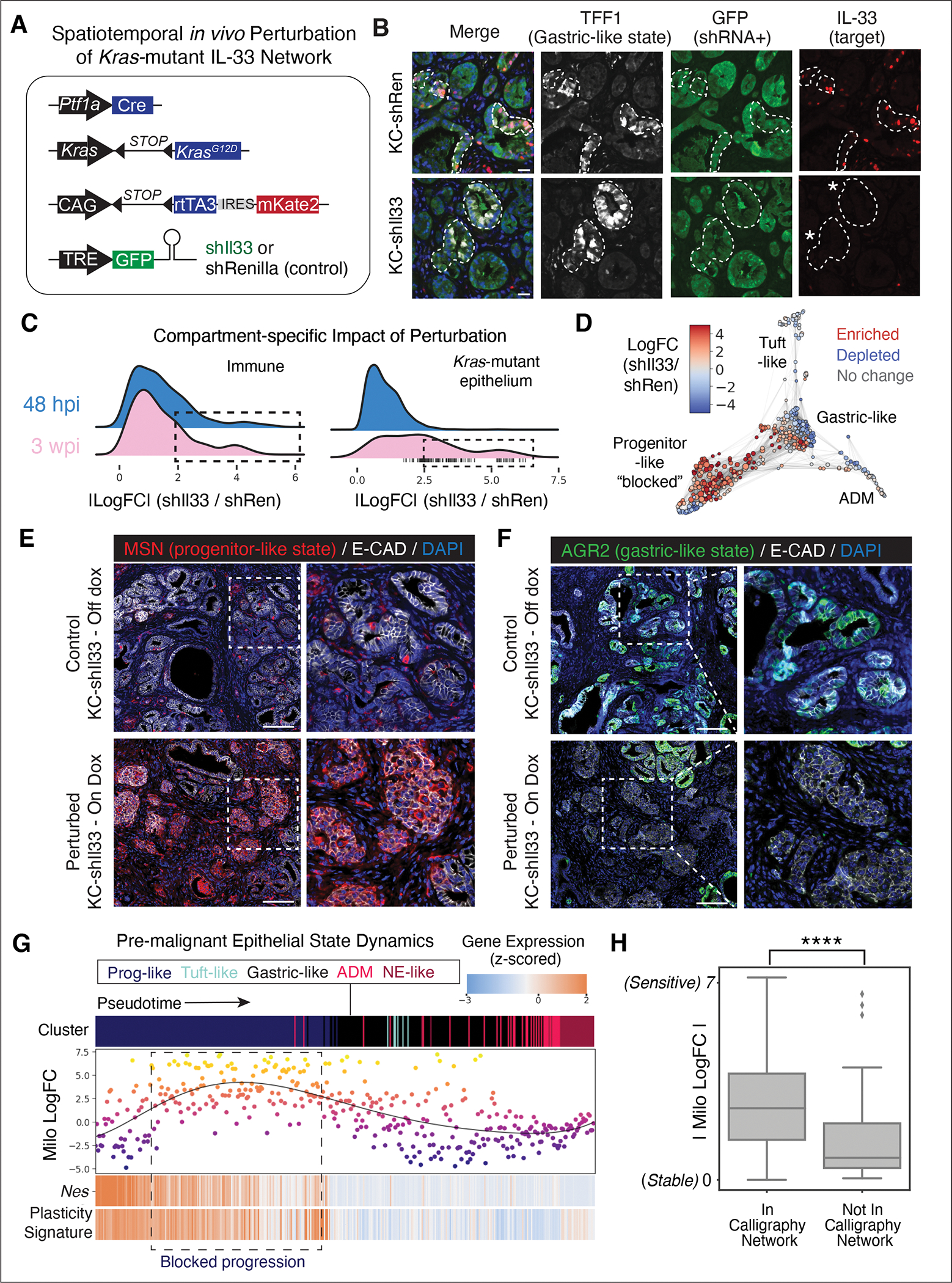
The response to tumor-initiating inflammatory and genetic insults can vary among morphologically indistinguishable cells, suggesting as yet uncharacterized roles for epigenetic plasticity during early neoplasia. To investigate the origins and impact of such plasticity, we performed single-cell analyses on normal, inflamed, premalignant, and malignant tissues in autochthonous models of pancreatic cancer. We reproducibly identified heterogeneous cell states that are primed for diverse, late-emerging neoplastic fates and linked these to chromatin remodeling at cell-cell communication loci. Using an inference approach, we revealed signaling gene modules and tissue-level cross-talk, including a neoplasia-driving feedback loop between discrete epithelial and immune cell populations that was functionally validated in mice. Our results uncover a neoplasia-specific tissue-remodeling program that may be exploited for pancreatic cancer interception.
Conflict of interest statement
Figures
Comment in
-
Digging deeper into the early steps of cancer progression.Trends Cancer. 2023 Sep;9(9):703-704. doi: 10.1016/j.trecan.2023.07.003. Epub 2023 Jul 25. Trends Cancer. 2023. PMID: 37495432
-
Calligraphy tool offers clues to the origin of pancreatic cancer.Nature. 2023 Sep;621(7978):265-266. doi: 10.1038/d41586-023-02518-4. Nature. 2023. PMID: 37587278 No abstract available.
References
-
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ, Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 348, 880–886 (2015). - PMC - PubMed
-
- Wijewardhane N, Dressler L, Ciccarelli FD, Normal Somatic Mutations in Cancer Transformation. Cancer Cell. 39, 125–129 (2021). - PubMed
-
- Hanahan D, Hallmarks of Cancer: New Dimensions. Cancer Discov. 12, 31–46 (2022). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
