

Bi-allelic variants in HMGCR cause an autosomal-recessive progressive limb-girdle muscular dystrophy
- PMID: 37167966
- PMCID: PMC10257193
- DOI: 10.1016/j.ajhg.2023.04.006
Bi-allelic variants in HMGCR cause an autosomal-recessive progressive limb-girdle muscular dystrophy
Abstract
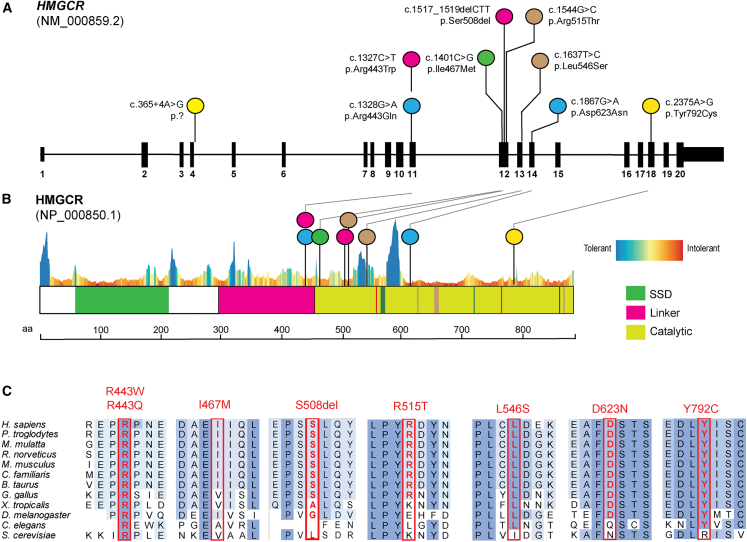
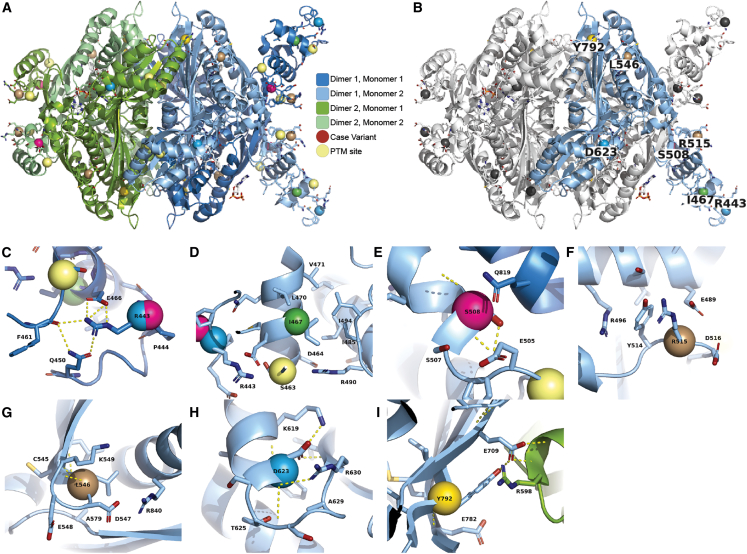
Statins are a mainstay intervention for cardiovascular disease prevention, yet their use can cause rare severe myopathy. HMG-CoA reductase, an essential enzyme in the mevalonate pathway, is the target of statins. We identified nine individuals from five unrelated families with unexplained limb-girdle like muscular dystrophy and bi-allelic variants in HMGCR via clinical and research exome sequencing. The clinical features resembled other genetic causes of muscular dystrophy with incidental high CPK levels (>1,000 U/L), proximal muscle weakness, variable age of onset, and progression leading to impaired ambulation. Muscle biopsies in most affected individuals showed non-specific dystrophic changes with non-diagnostic immunohistochemistry. Molecular modeling analyses revealed variants to be destabilizing and affecting protein oligomerization. Protein activity studies using three variants (p.Asp623Asn, p.Tyr792Cys, and p.Arg443Gln) identified in affected individuals confirmed decreased enzymatic activity and reduced protein stability. In summary, we showed that individuals with bi-allelic amorphic (i.e., null and/or hypomorphic) variants in HMGCR display phenotypes that resemble non-genetic causes of myopathy involving this reductase. This study expands our knowledge regarding the mechanisms leading to muscular dystrophy through dysregulation of the mevalonate pathway, autoimmune myopathy, and statin-induced myopathy.
Keywords: HMG-CoA reducatase; HMGCR; autoimmune myopathy; limb-girdle like muscular dystrophy; mevalonate pathway; rare genetic disease; statin-induced myopathy.
Copyright © 2023. Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing from Baylor Genetics Laboratories. J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting.
Figures



Similar articles
-
Limb girdle muscular disease caused by HMGCR mutation and statin myopathy treatable with mevalonolactone.Proc Natl Acad Sci U S A. 2023 Feb 14;120(7):e2217831120. doi: 10.1073/pnas.2217831120. Epub 2023 Feb 6. Proc Natl Acad Sci U S A. 2023. PMID: 36745799 Free PMC article.
-
Slowly Progressive Limb-Girdle Weakness and HyperCKemia - Limb Girdle Muscular Dystrophy or Anti-3-Hydroxy-3-Methylglutaryl-CoA-Reductase-Myopathy?J Neuromuscul Dis. 2022;9(5):607-614. doi: 10.3233/JND-220810. J Neuromuscul Dis. 2022. PMID: 35754285
-
Anti-HMGCR myopathy may resemble limb-girdle muscular dystrophy.Neurol Neuroimmunol Neuroinflamm. 2018 Dec 12;6(1):e523. doi: 10.1212/NXI.0000000000000523. eCollection 2019 Jan. Neurol Neuroimmunol Neuroinflamm. 2018. PMID: 30588482 Free PMC article.
-
Anti-HMGCR Myopathy.J Neuromuscul Dis. 2018;5(1):11-20. doi: 10.3233/JND-170282. J Neuromuscul Dis. 2018. PMID: 29480216 Free PMC article. Review.
-
Update on the genetics of limb girdle muscular dystrophy.Semin Pediatr Neurol. 2012 Dec;19(4):211-8. doi: 10.1016/j.spen.2012.09.008. Semin Pediatr Neurol. 2012. PMID: 23245554 Review.
Cited by
-
Anti-Ku + myositis: an acquired inflammatory protein-aggregate myopathy.Acta Neuropathol. 2024 Jul 16;148(1):6. doi: 10.1007/s00401-024-02765-3. Acta Neuropathol. 2024. PMID: 39012547 Free PMC article.
-
Expert panel curation of 31 genes in relation to limb girdle muscular dystrophy.Ann Clin Transl Neurol. 2024 Sep;11(9):2268-2276. doi: 10.1002/acn3.52127. Epub 2024 Aug 30. Ann Clin Transl Neurol. 2024. PMID: 39215466 Free PMC article.
-
High-Density Lipoprotein-Associated Cholesterol Abnormalities in a Clinical Outcomes Study of Dysferlin-Deficient Limb-Girdle Muscular Dystrophy Type R2.J Cachexia Sarcopenia Muscle. 2025 Aug;16(4):e70042. doi: 10.1002/jcsm.70042. J Cachexia Sarcopenia Muscle. 2025. PMID: 40814795 Free PMC article.
-
Pathogenic autoantibody internalization in myositis.medRxiv [Preprint]. 2024 Jan 17:2024.01.15.24301339. doi: 10.1101/2024.01.15.24301339. medRxiv. 2024. Update in: Ann Rheum Dis. 2024 Oct 21;83(11):1549-1560. doi: 10.1136/ard-2024-225773. PMID: 38313303 Free PMC article. Updated. Preprint.
-
Drosophila melanogaster: A Model Organism in Muscular Dystrophy Studies.Int J Mol Sci. 2025 Feb 10;26(4):1459. doi: 10.3390/ijms26041459. Int J Mol Sci. 2025. PMID: 40003927 Free PMC article. Review.
References
-
- Reddy H.M., Cho K.A., Lek M., Estrella E., Valkanas E., Jones M.D., Mitsuhashi S., Darras B.T., Amato A.A., Lidov H.G., et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J. Hum. Genet. 2017;62:243–252. doi: 10.1038/jhg.2016.116. - DOI - PMC - PubMed
-
- Töpf A., Johnson K., Bates A., Phillips L., Chao K.R., England E.M., Laricchia K.M., Mullen T., Valkanas E., Xu L., et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. 2020;22:1478–1488. doi: 10.1038/s41436-020-0840-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
