

Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant
- PMID: 37169744
- PMCID: PMC10175283
- DOI: 10.1038/s41467-023-38188-z
Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant
Abstract
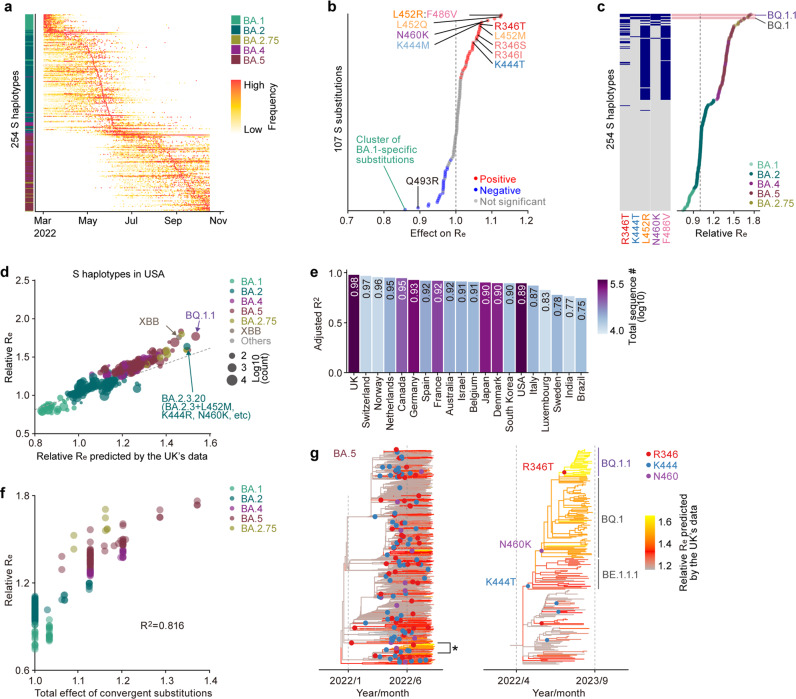
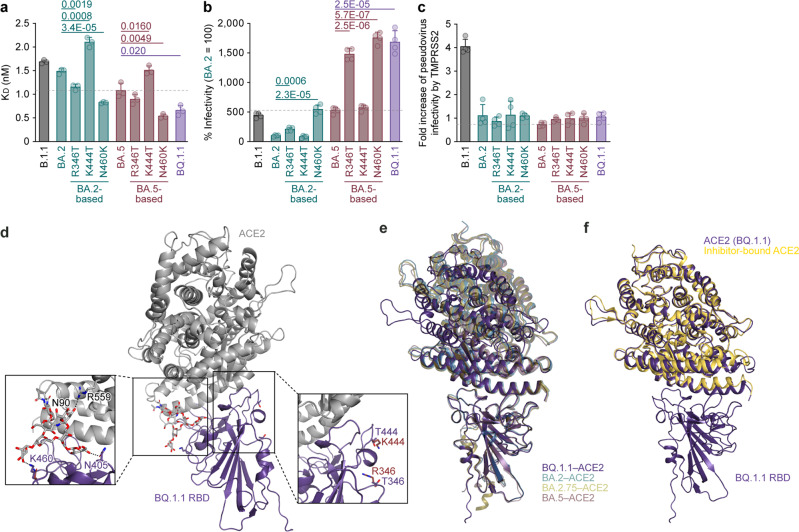
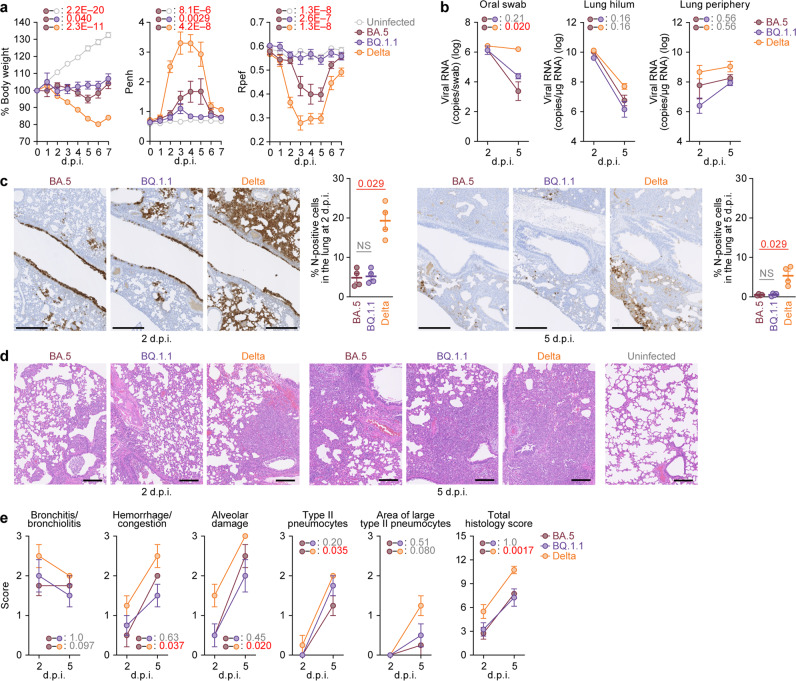
In late 2022, various Omicron subvariants emerged and cocirculated worldwide. These variants convergently acquired amino acid substitutions at critical residues in the spike protein, including residues R346, K444, L452, N460, and F486. Here, we characterize the convergent evolution of Omicron subvariants and the properties of one recent lineage of concern, BQ.1.1. Our phylogenetic analysis suggests that these five substitutions are recurrently acquired, particularly in younger Omicron lineages. Epidemic dynamics modelling suggests that the five substitutions increase viral fitness, and a large proportion of the fitness variation within Omicron lineages can be explained by these substitutions. Compared to BA.5, BQ.1.1 evades breakthrough BA.2 and BA.5 infection sera more efficiently, as demonstrated by neutralization assays. The pathogenicity of BQ.1.1 in hamsters is lower than that of BA.5. Our multiscale investigations illuminate the evolutionary rules governing the convergent evolution for known Omicron lineages as of 2022.
© 2023. The Author(s).
Conflict of interest statement
Y.Y. and T.N. are founders and shareholders of HiLung, Inc. Y.Y. is a coinventor of patents (PCT/JP2016/057254; “Method for inducing differentiation of alveolar epithelial cells”, PCT/JP2016/059786, “Method of producing airway epithelial cells”). The other authors declare that no competing interests exist.
Figures



References
-
- WHO. Tracking SARS-CoV-2 variants (March 30, 2023) https://www.who.int/en/activities/tracking-SARS-CoV-2-variants (2022).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
