

Comparative analysis of cell-cell communication at single-cell resolution
- PMID: 37169965
- PMCID: PMC10638471
- DOI: 10.1038/s41587-023-01782-z
Comparative analysis of cell-cell communication at single-cell resolution
Abstract
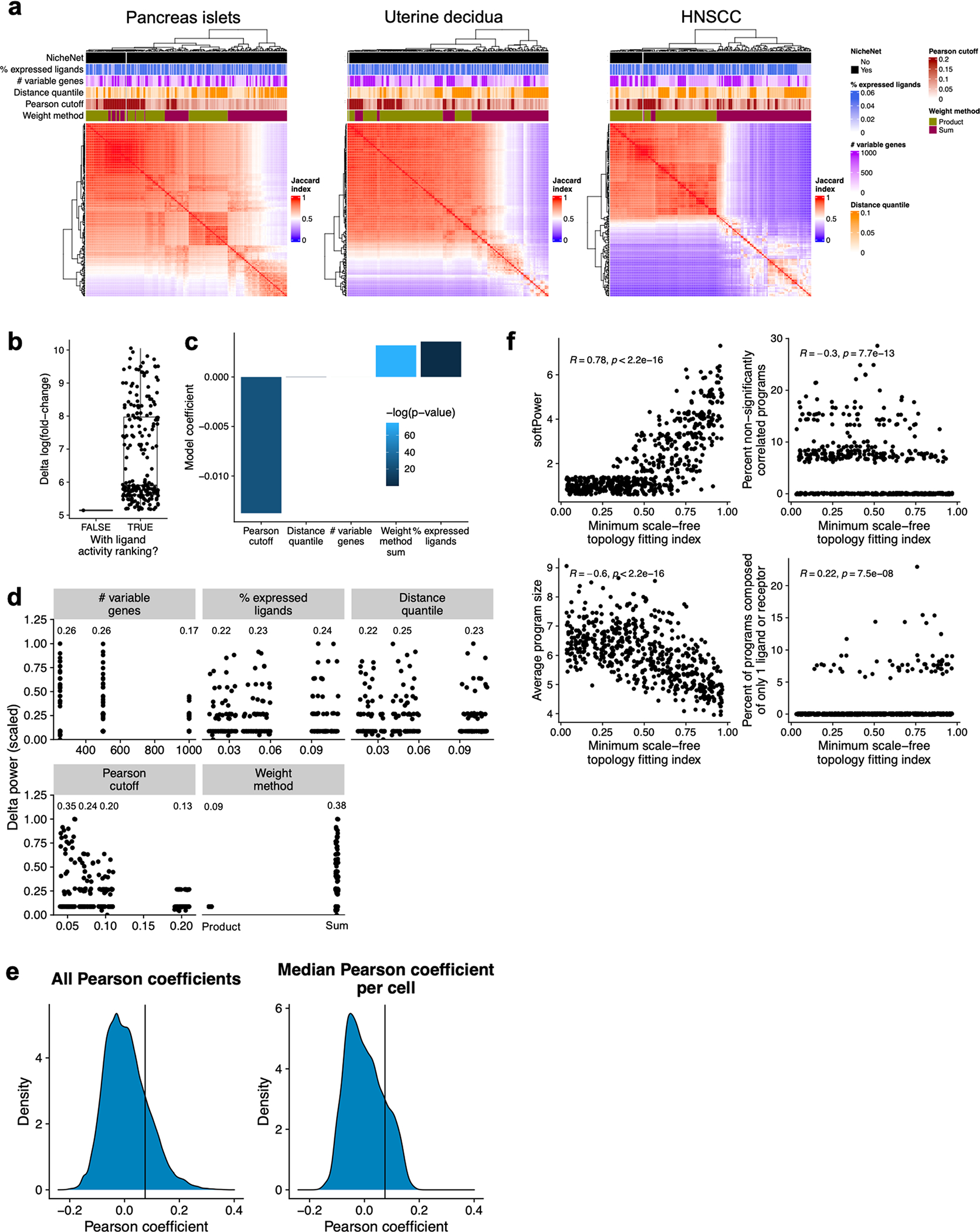
Inference of cell-cell communication from single-cell RNA sequencing data is a powerful technique to uncover intercellular communication pathways, yet existing methods perform this analysis at the level of the cell type or cluster, discarding single-cell-level information. Here we present Scriabin, a flexible and scalable framework for comparative analysis of cell-cell communication at single-cell resolution that is performed without cell aggregation or downsampling. We use multiple published atlas-scale datasets, genetic perturbation screens and direct experimental validation to show that Scriabin accurately recovers expected cell-cell communication edges and identifies communication networks that can be obscured by agglomerative methods. Additionally, we use spatial transcriptomic data to show that Scriabin can uncover spatial features of interaction from dissociated data alone. Finally, we demonstrate applications to longitudinal datasets to follow communication pathways operating between timepoints. Our approach represents a broadly applicable strategy to reveal the full structure of niche-phenotype relationships in health and disease.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
A.K.S. reports compensation for consulting and/or scientific advisory board (SAB) membership from Merck, Honeycomb Biotechnologies, Cellarity, Repertoire Immune Medicines, Ochre Bio, Third Rock Ventures, Hovione, Relation Therapeutics, FL82, Empress Therapeutics, IntrECate Biotherapeutics, Senda Biosciences and Dahlia Biosciences. C.A.B. reports compensation for consulting and/or SAB membership from Catamaran Bio, DeepCell, Immunebridge and Revelation Biosciences. The remaining authors declare no competing interests.
Figures















Update of
-
Comparative analysis of cell-cell communication at single-cell resolution.bioRxiv [Preprint]. 2022 Jul 25:2022.02.04.479209. doi: 10.1101/2022.02.04.479209. bioRxiv. 2022. Update in: Nat Biotechnol. 2024 Mar;42(3):470-483. doi: 10.1038/s41587-023-01782-z. PMID: 35169794 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
