

A prime editor mouse to model a broad spectrum of somatic mutations in vivo
- PMID: 37169967
- PMCID: PMC11120832
- DOI: 10.1038/s41587-023-01783-y
A prime editor mouse to model a broad spectrum of somatic mutations in vivo
Abstract
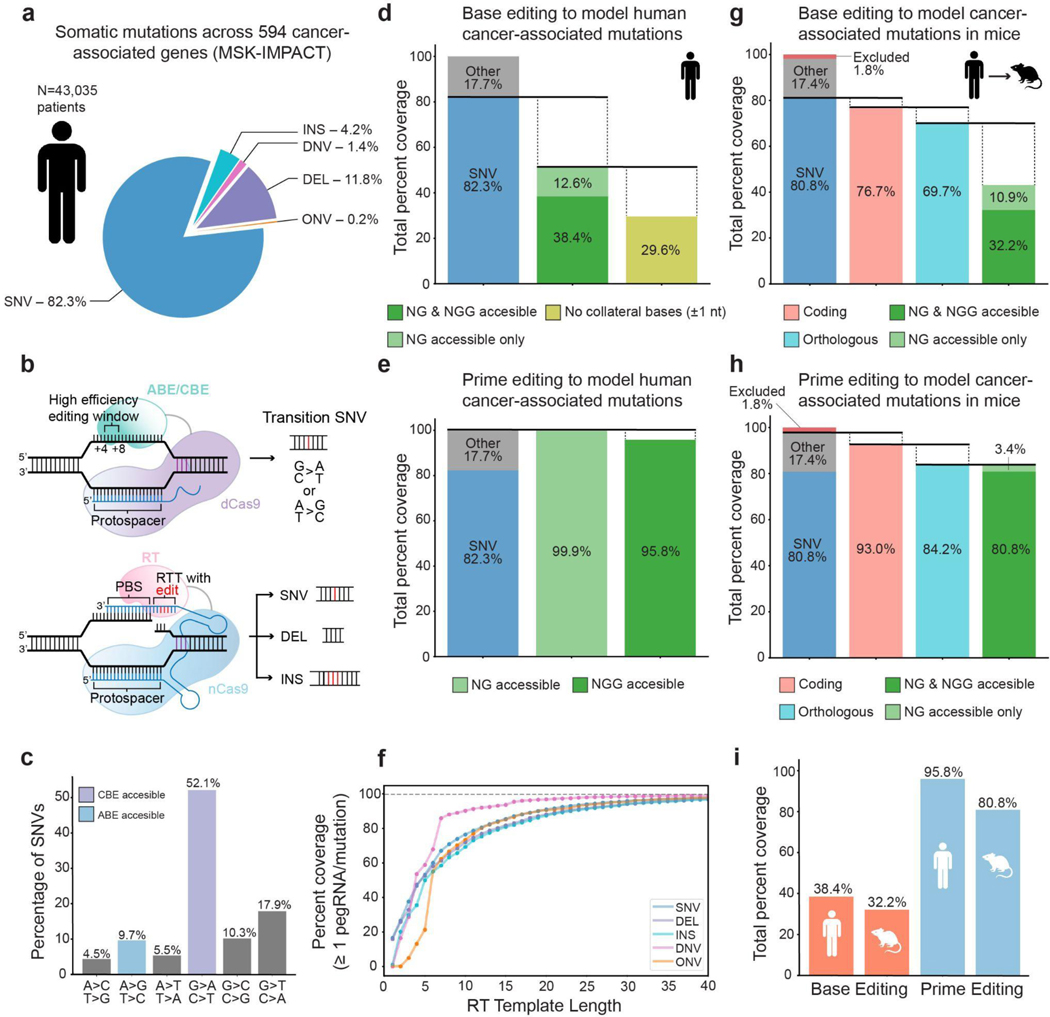
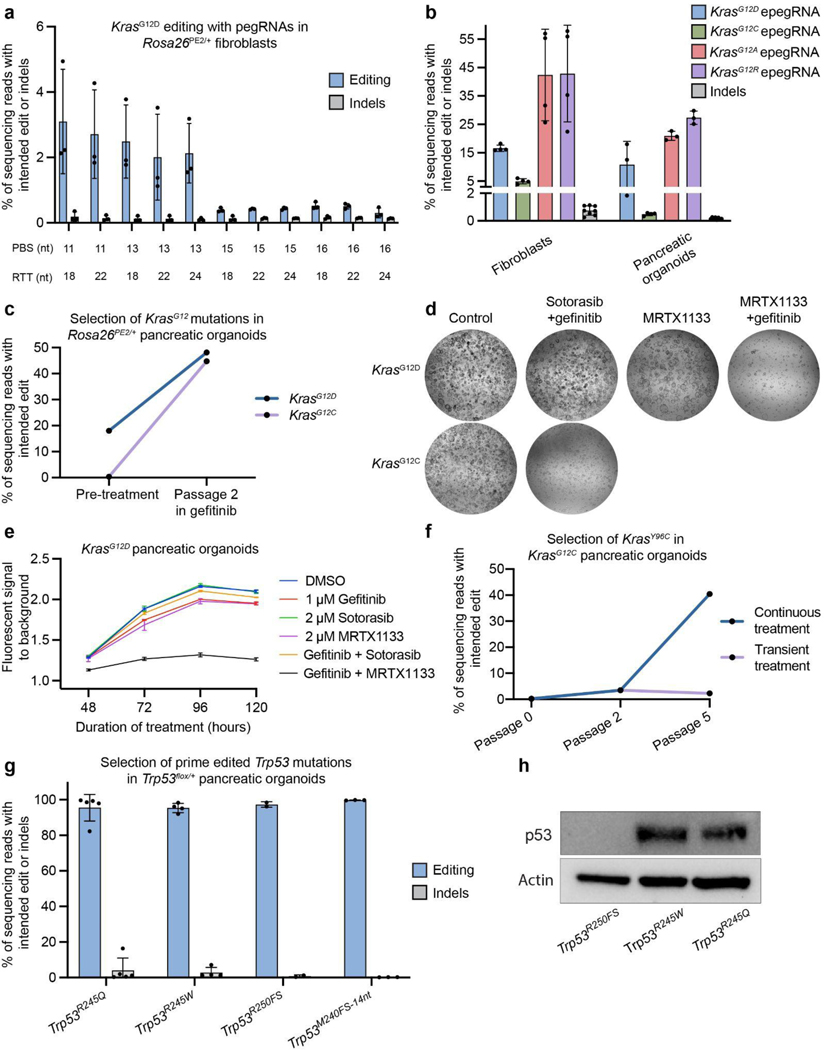
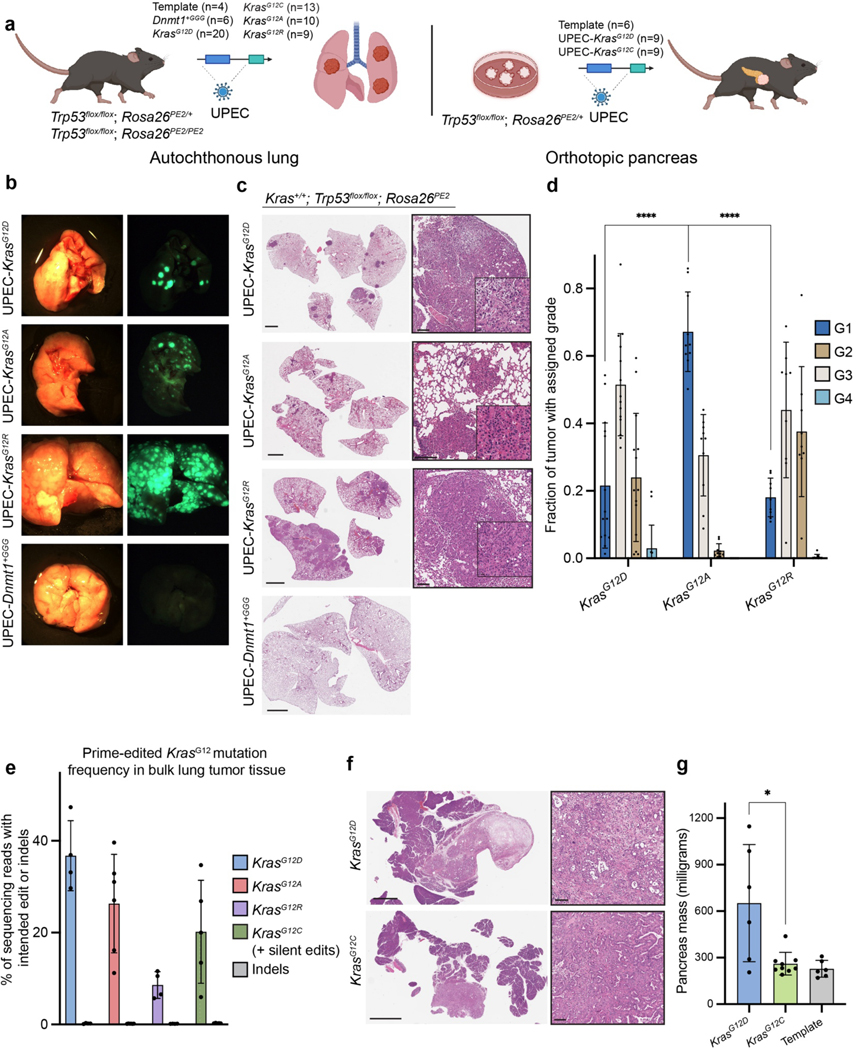
Genetically engineered mouse models only capture a small fraction of the genetic lesions that drive human cancer. Current CRISPR-Cas9 models can expand this fraction but are limited by their reliance on error-prone DNA repair. Here we develop a system for in vivo prime editing by encoding a Cre-inducible prime editor in the mouse germline. This model allows rapid, precise engineering of a wide range of mutations in cell lines and organoids derived from primary tissues, including a clinically relevant Kras mutation associated with drug resistance and Trp53 hotspot mutations commonly observed in pancreatic cancer. With this system, we demonstrate somatic prime editing in vivo using lipid nanoparticles, and we model lung and pancreatic cancer through viral delivery of prime editing guide RNAs or orthotopic transplantation of prime-edited organoids. We believe that this approach will accelerate functional studies of cancer-associated mutations and complex genetic combinations that are challenging to construct with traditional models.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Declaration of Interests:
T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. T.J. is also the President of Break Through Cancer. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. His laboratory currently receives funding from the Johnson & Johnson Lung Cancer Initiative, but this funding did not support the research described in this manuscript. D.R.L. is a consultant for Prime Medicine, Beam Therapeutics, Pairwise Plants, Chroma Medicine, and Nvelop Therapeutics, companies that use or deliver genome editing or genome engineering agents and owns equity in these companies. K.H. and A.V.A. are currently employees of Prime Medicine.
Figures

References
-
- Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37 (2013). - PubMed
MeSH terms
Substances
Grants and funding
- T32 GM007287/GM/NIGMS NIH HHS/United States
- K99 HL163805/HL/NHLBI NIH HHS/United States
- RM1 HG009490/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R35 GM118062/GM/NIGMS NIH HHS/United States
- T32 GM144273/GM/NIGMS NIH HHS/United States
- T32 GM136540/GM/NIGMS NIH HHS/United States
- U01 AI142756/AI/NIAID NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- K08 CA259621/CA/NCI NIH HHS/United States
- F31 CA268835/CA/NCI NIH HHS/United States
- P30 CA014051/CA/NCI NIH HHS/United States
- R00 HL163805/HL/NHLBI NIH HHS/United States
- K22 CA279501/CA/NCI NIH HHS/United States
- T32 CA009172/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
