

Animal Models of Drug-Resistant Epilepsy as Tools for Deciphering the Cellular and Molecular Mechanisms of Pharmacoresistance and Discovering More Effective Treatments
- PMID: 37174633
- PMCID: PMC10177106
- DOI: 10.3390/cells12091233
Animal Models of Drug-Resistant Epilepsy as Tools for Deciphering the Cellular and Molecular Mechanisms of Pharmacoresistance and Discovering More Effective Treatments
Abstract
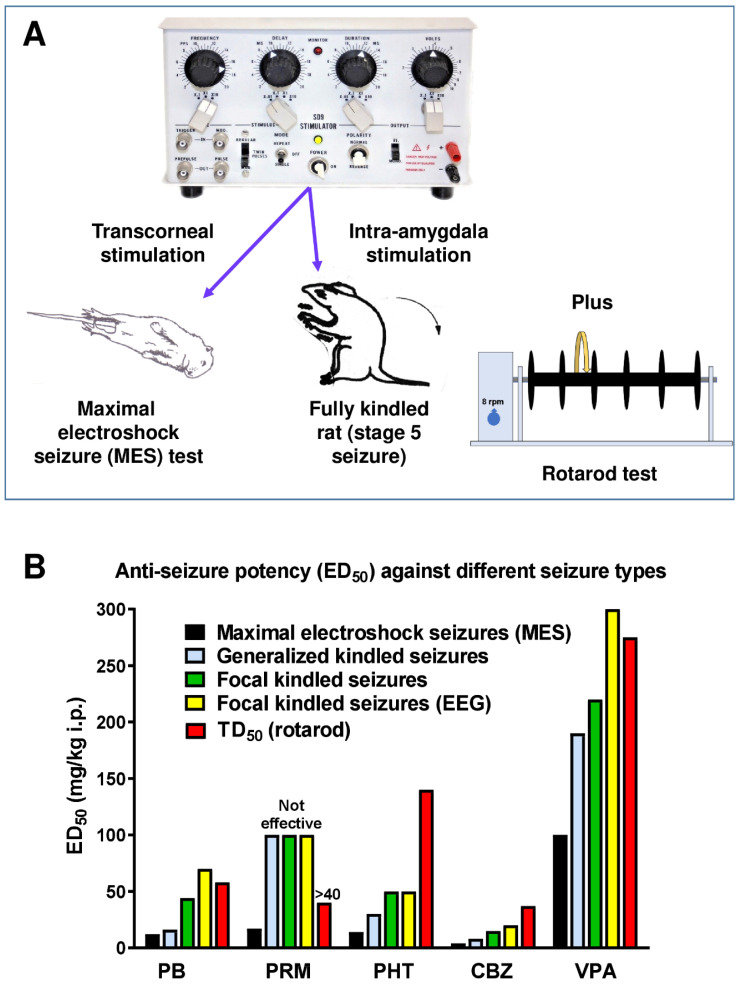
In the last 30 years, over 20 new anti-seizure medicines (ASMs) have been introduced into the market for the treatment of epilepsy using well-established preclinical seizure and epilepsy models. Despite this success, approximately 20-30% of patients with epilepsy have drug-resistant epilepsy (DRE). The current approach to ASM discovery for DRE relies largely on drug testing in various preclinical model systems that display varying degrees of ASM drug resistance. In recent years, attempts have been made to include more etiologically relevant models in the preclinical evaluation of a new investigational drug. Such models have played an important role in advancing a greater understanding of DRE at a mechanistic level and for hypothesis testing as new experimental evidence becomes available. This review provides a critical discussion of the pharmacology of models of adult focal epilepsy that allow for the selection of ASM responders and nonresponders and those models that display a pharmacoresistance per se to two or more ASMs. In addition, the pharmacology of animal models of major genetic epilepsies is discussed. Importantly, in addition to testing chemical compounds, several of the models discussed here can be used to evaluate other potential therapies for epilepsy such as neurostimulation, dietary treatments, gene therapy, or cell transplantation. This review also discusses the challenges associated with identifying novel therapies in the absence of a greater understanding of the mechanisms that contribute to DRE. Finally, this review discusses the lessons learned from the profile of the recently approved highly efficacious and broad-spectrum ASM cenobamate.
Keywords: Dravet syndrome; anti-seizure drugs; drug resistant epilepsy; kainate; kindling; pilocarpine; seizures; temporal lobe epilepsy.
Conflict of interest statement
W. Löscher was involved in the development of levetiracetam (UCB Pharma; Brussels, Belgium) and imepitoin (Elbion/Boehringer Ingelheim, Germany), has received consultancy fees from Lundbeck, Angelini, AC Immune, Clexio Biosciences, UCB Pharma, Pragma Therapeutics, Boehringer Ingelheim, Pfizer, and Johnson & Johnson, and has served on the advisory boards of Grünenthal, UCB Pharma, and Angelini Pharma. He is a member of the external consultant board (ECB) of the Epilepsy Therapy Screening Program (ETSP) of the National Institute of Neurological Disorders and Stroke, National Institutes of Health, Rockville, MD, USA. He is also co-founder of PrevEp Inc., Bethesda, MD. H.S. White has received grant funding from UCB Pharma and Eisai Pharmaceuticals; consultant fees from Biogen, GW, Neurelis, Inc. Takeda, Inc. and JAZZ Pharmaceuticals; and speaker honoraria from SK Pharmaceuticals and UCB Pharma. H.S. White is also co-founder of NeuroAdjuvants, Inc., Salt Lake City, UT, USA.
Figures









References
-
- Galanopoulou A.S., Buckmaster P.S., Staley K.J., Moshé S.L., Perucca E., Engel J., Jr., Löscher W., Noebels J.L., Pitkänen A., Stables J., et al. The American Epilepsy Society Basic Science Committee and The International League Against Epilepsy Working Group on Recommendations for Preclinical Epilepsy Drug Discovery. Identification of new epilepsy treatments: Issues in preclinical methodology. Epilepsia. 2012;53:571–582. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
