

Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates
- PMID: 37175085
- PMCID: PMC10180314
- DOI: 10.3390/molecules28093675
Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates
Abstract
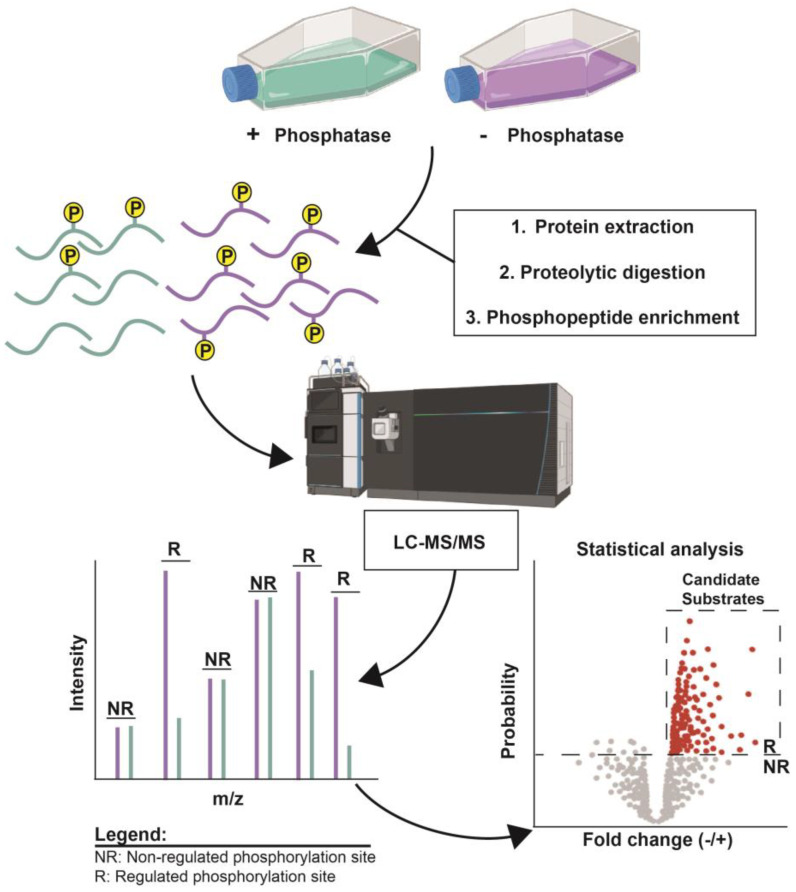
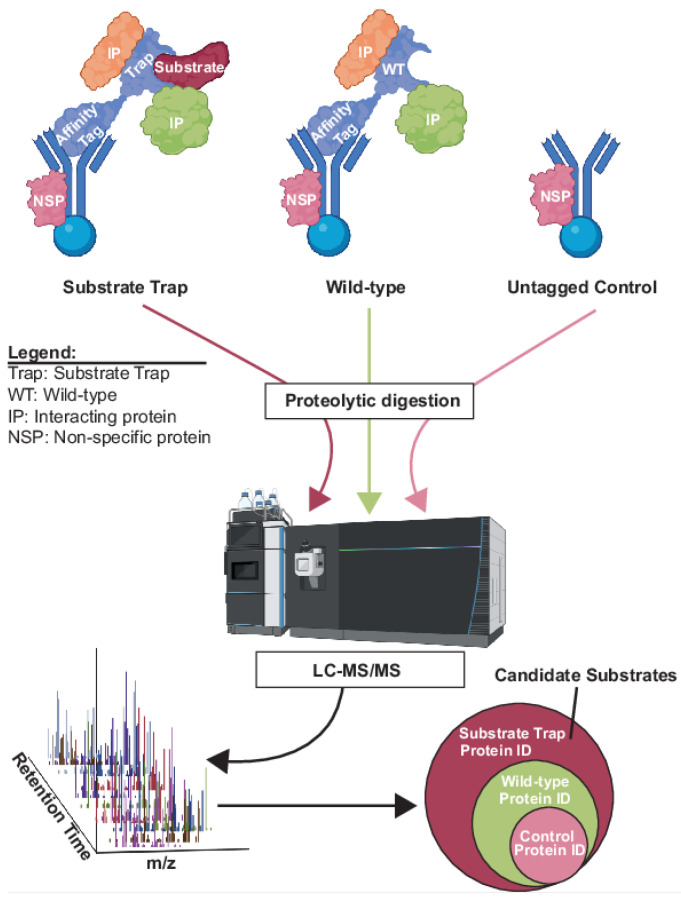
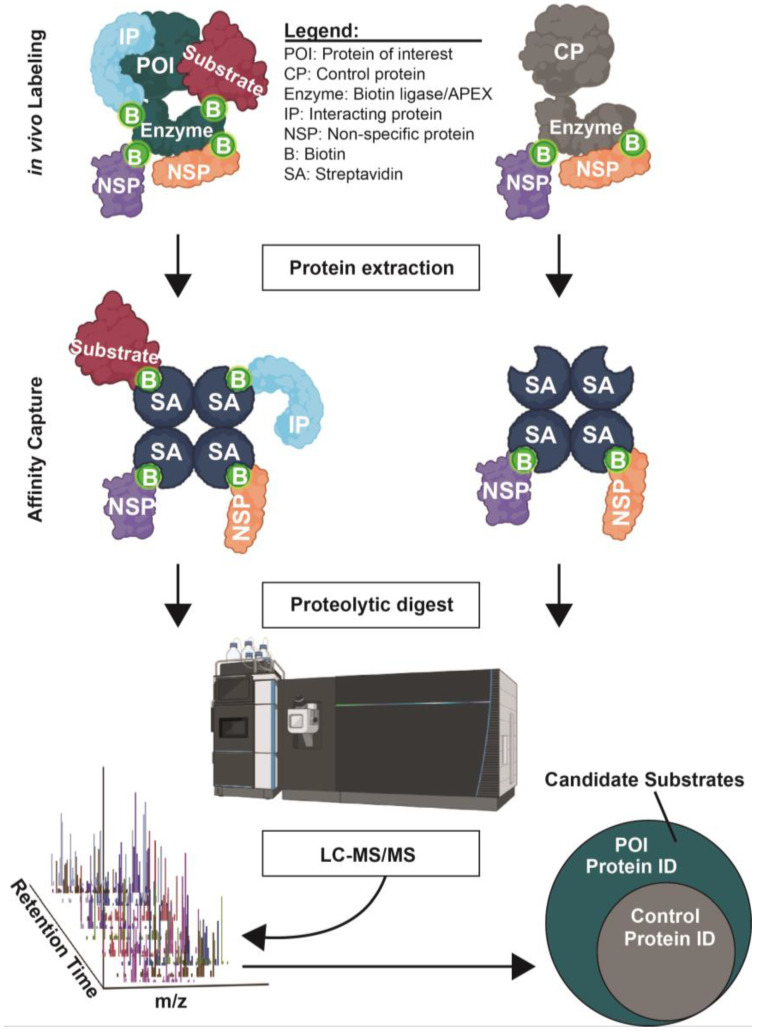
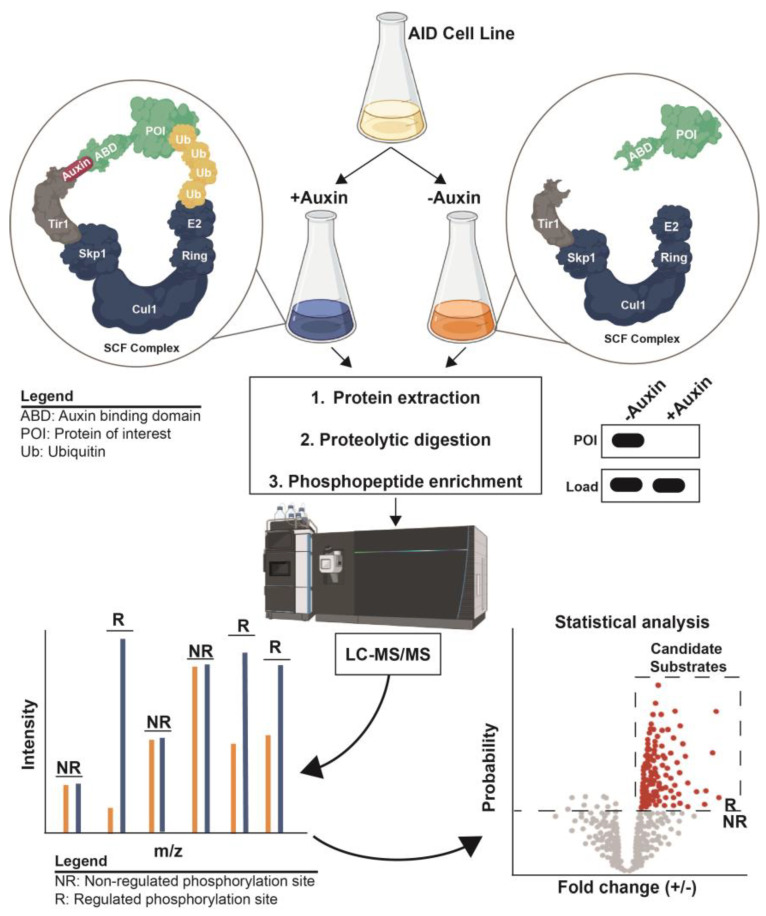
Protein phosphorylation is a ubiquitous post-translational modification controlled by the opposing activities of protein kinases and phosphatases, which regulate diverse biological processes in all kingdoms of life. One of the key challenges to a complete understanding of phosphoregulatory networks is the unambiguous identification of kinase and phosphatase substrates. Liquid chromatography-coupled mass spectrometry (LC-MS/MS) and associated phosphoproteomic tools enable global surveys of phosphoproteome changes in response to signaling events or perturbation of phosphoregulatory network components. Despite the power of LC-MS/MS, it is still challenging to directly link kinases and phosphatases to specific substrate phosphorylation sites in many experiments. Here, we survey common LC-MS/MS-based phosphoproteomic workflows for identifying protein kinase and phosphatase substrates, noting key advantages and limitations of each. We conclude by discussing the value of inducible degradation technologies coupled with phosphoproteomics as a new approach that overcomes some limitations of current methods for substrate identification of kinases, phosphatases, and other regulatory enzymes.
Keywords: PTM; kinase; mass spectrometry; phosphatase; phosphoproteomics; phosphorylation; post-translation modification; quantitative proteomics; substrate identification.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The coming of age of phosphoproteomics--from large data sets to inference of protein functions.Mol Cell Proteomics. 2013 Dec;12(12):3453-64. doi: 10.1074/mcp.R113.032862. Epub 2013 Sep 13. Mol Cell Proteomics. 2013. PMID: 24037665 Free PMC article. Review.
-
Phosphatase and Kinase Substrate Specificity Profiling with Pooled Synthetic Peptides and Mass Spectrometry.Methods Mol Biol. 2021;2329:51-70. doi: 10.1007/978-1-0716-1538-6_5. Methods Mol Biol. 2021. PMID: 34085215
-
Abundant protein phosphorylation potentially regulates Arabidopsis anther development.J Exp Bot. 2016 Sep;67(17):4993-5008. doi: 10.1093/jxb/erw293. Epub 2016 Aug 16. J Exp Bot. 2016. PMID: 27531888 Free PMC article.
-
Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast.Sci Signal. 2010 Dec 21;3(153):rs4. doi: 10.1126/scisignal.2001182. Sci Signal. 2010. PMID: 21177495 Free PMC article.
-
Quantitative phosphoproteomics strategies for understanding protein kinase-mediated signal transduction pathways.Expert Rev Proteomics. 2011 Feb;8(1):81-94. doi: 10.1586/epr.10.104. Expert Rev Proteomics. 2011. PMID: 21329429 Review.
Cited by
-
The Fascinating Intricacy of pSer/Thr-Specific Phosphatases and Their Higher-Order Complexes: Emerging Concepts.Biochemistry. 2025 Jun 17;64(12):2506-2515. doi: 10.1021/acs.biochem.5c00183. Epub 2025 Jun 5. Biochemistry. 2025. PMID: 40473240 Review.
-
MAP2 phosphorylation: mechanisms, functional consequences, and emerging insights.Front Cell Neurosci. 2025 Jul 30;19:1610371. doi: 10.3389/fncel.2025.1610371. eCollection 2025. Front Cell Neurosci. 2025. PMID: 40810115 Free PMC article. Review.
-
The Hunt Lab Weighs in on Mass Spectrometry-Based Analysis of Protein Posttranslational Modifications.Mol Cell Proteomics. 2025 Apr;24(4):100943. doi: 10.1016/j.mcpro.2025.100943. Epub 2025 Mar 11. Mol Cell Proteomics. 2025. PMID: 40081537 Free PMC article. Review.
-
Inducible degradation-coupled phosphoproteomics identifies PP2ARts1 as a novel eisosome regulator.Front Cell Dev Biol. 2024 Aug 21;12:1451027. doi: 10.3389/fcell.2024.1451027. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39234563 Free PMC article.
-
PP2A-B56α is a key determinant of cardiac protein phosphorylation and functional responses to β-adrenergic signalling.J Mol Cell Cardiol Plus. 2025 May 3;12:100301. doi: 10.1016/j.jmccpl.2025.100301. eCollection 2025 Jun. J Mol Cell Cardiol Plus. 2025. PMID: 40485773 Free PMC article.
References
-
- Mertins P., Tang L.C., Krug K., Clark D.J., Gritsenko M.A., Chen L., Clauser K.R., Clauss T.R., Shah P., Gillette M.A., et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat. Protoc. 2018;13:1632–1661. doi: 10.1038/s41596-018-0006-9. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
