

Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum-Classical Vibronic Approaches-Application to Coumarin C153 in Methanol
- PMID: 37175320
- PMCID: PMC10180259
- DOI: 10.3390/molecules28093910
Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum-Classical Vibronic Approaches-Application to Coumarin C153 in Methanol
Abstract
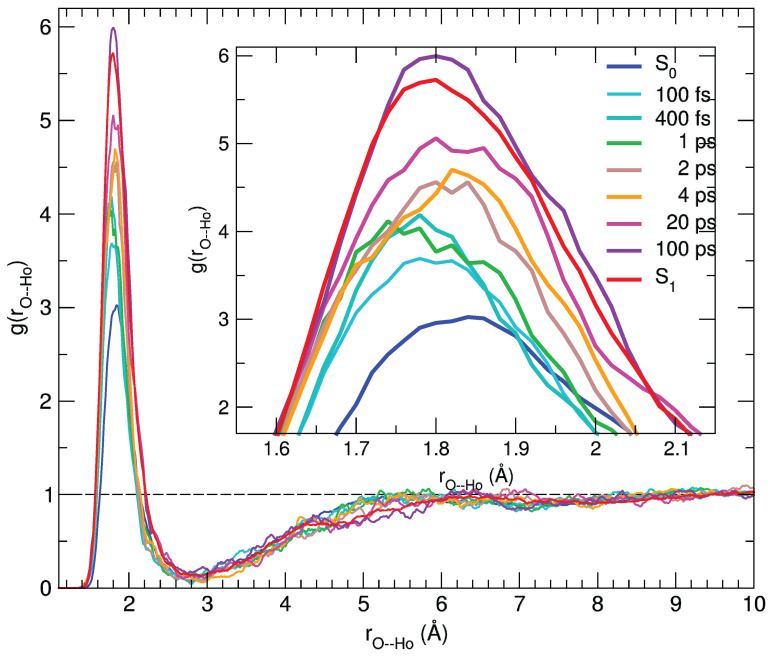
We report a joint experimental and theoretical work on the steady-state spectroscopy and time-resolved emission of the coumarin C153 dye in methanol. The lowest energy excited state of this molecule is characterized by an intramolecular charge transfer thus leading to remarkable shifts of the time-resolved emission spectra, dictated by the methanol reorganization dynamics. We selected this system as a prototypical test case for the first application of a novel computational protocol aimed at the prediction of transient emission spectral shapes, including both vibronic and solvent effects, without applying any phenomenological broadening. It combines a recently developed quantum-classical approach, the adiabatic molecular dynamics generalized vertical Hessian method (Ad-MD|gVH), with nonequilibrium molecular dynamics simulations. For the steady-state spectra we show that the Ad-MD|gVH approach is able to reproduce quite accurately the spectral shapes and the Stokes shift, while a ∼0.15 eV error is found on the prediction of the solvent shift going from gas phase to methanol. The spectral shape of the time-resolved emission signals is, overall, well reproduced, although the simulated spectra are slightly too broad and asymmetric at low energies with respect to experiments. As far as the spectral shift is concerned, the calculated spectra from 4 ps to 100 ps are in excellent agreement with experiments, correctly predicting the end of the solvent reorganization after about 20 ps. On the other hand, before 4 ps solvent dynamics is predicted to be too fast in the simulations and, in the sub-ps timescale, the uncertainty due to the experimental time resolution (300 fs) makes the comparison less straightforward. Finally, analysis of the reorganization of the first solvation shell surrounding the excited solute, based on atomic radial distribution functions and orientational correlations, indicates a fast solvent response (≈100 fs) characterized by the strengthening of the carbonyl-methanol hydrogen bond interactions, followed by the solvent reorientation, occurring on the ps timescale, to maximize local dipolar interactions.
Keywords: force field parameterization; mixed quantum chemical approaches; molecular dynamics; organic dyes; time-dependent density functional theory; time-resolved emission spectroscopy; vibronic approaches.
Conflict of interest statement
The authors declare no conflict of interest.
Figures







References
-
- He Y., Tang L., Wu X., Hou X., Lee Y.I. Spectroscopy: The best way toward green analytical chemistry? Appl. Spectrosc. Rev. 2007;42:119–138. doi: 10.1080/05704920601184259. - DOI
-
- Laane J. Frontiers of Molecular Spectroscopy. Elsevier; Amsterdam, The Netherlands: 2011.
-
- Sasaki S., Drummen G.P., Konishi G.i. Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C. 2016;4:2731–2743. doi: 10.1039/C5TC03933A. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
