

Sites of transcription initiation drive mRNA isoform selection
- PMID: 37178687
- PMCID: PMC10228280
- DOI: 10.1016/j.cell.2023.04.012
Sites of transcription initiation drive mRNA isoform selection
Abstract
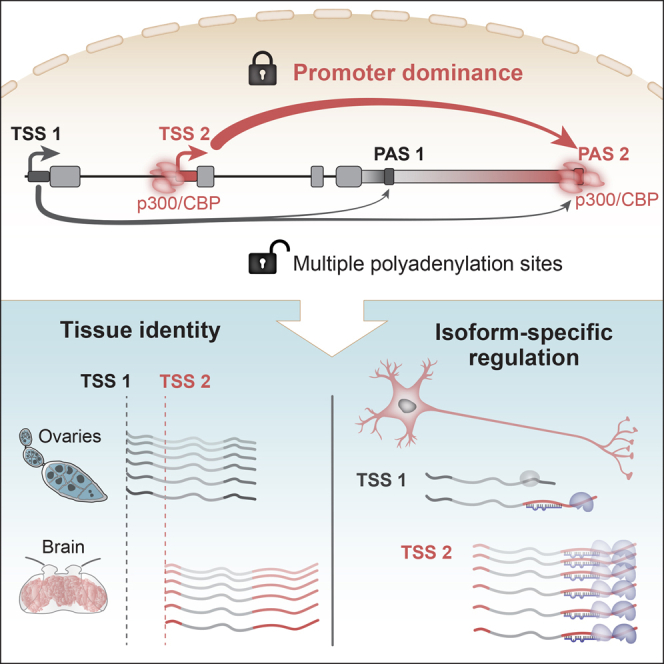
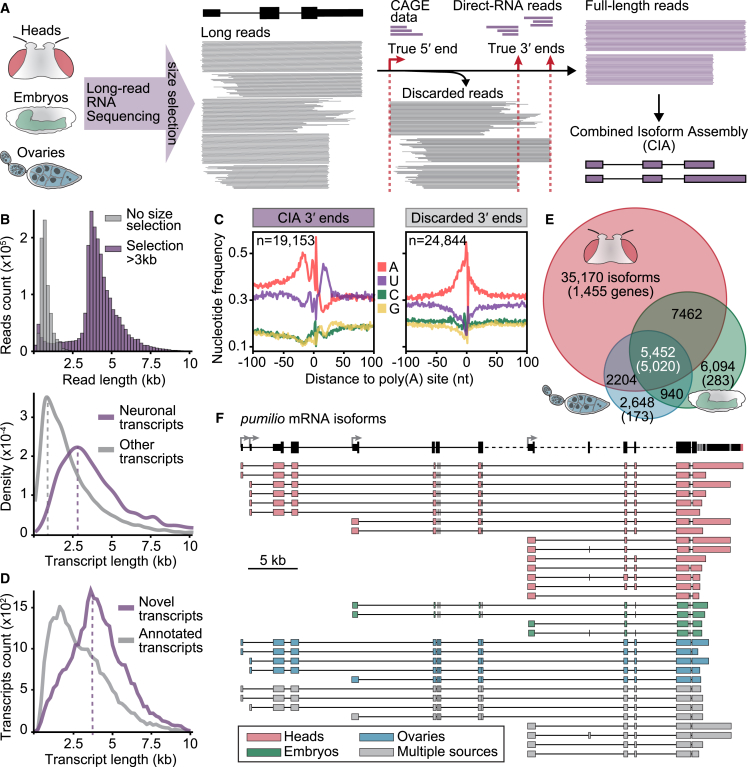
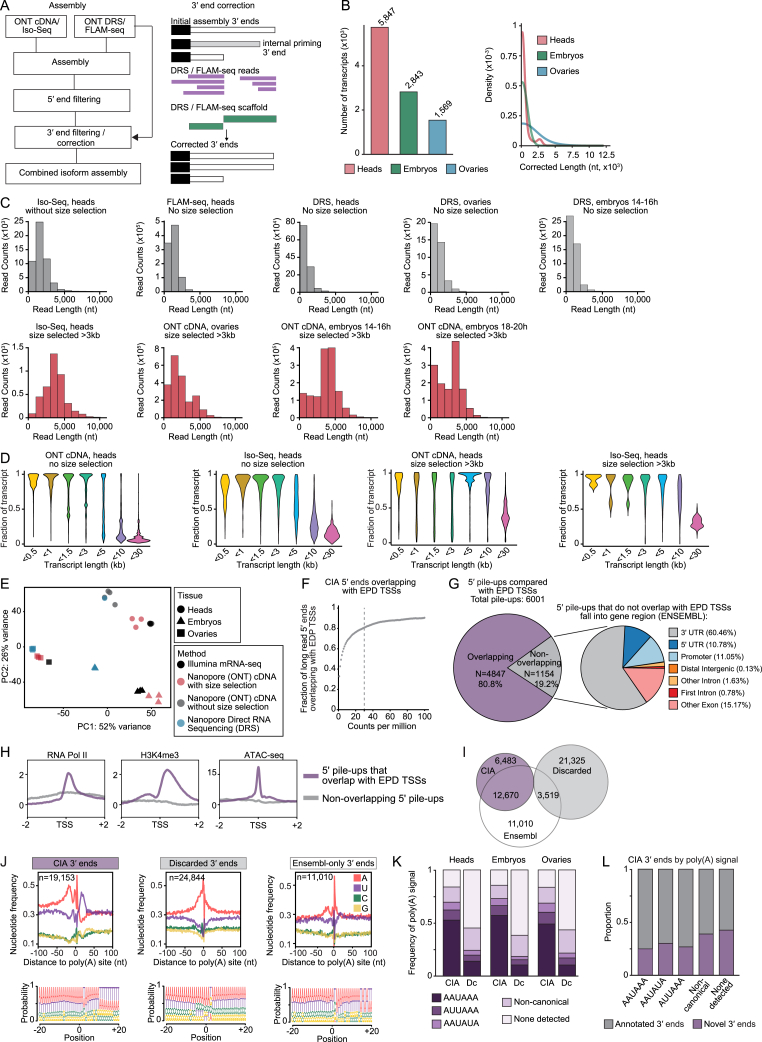
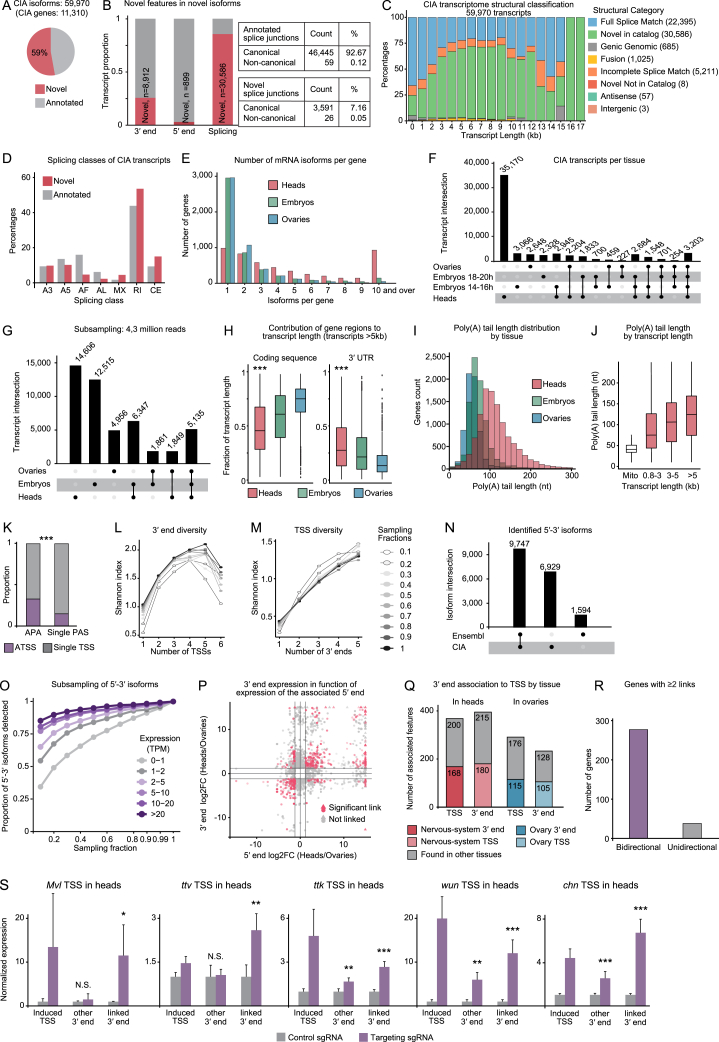
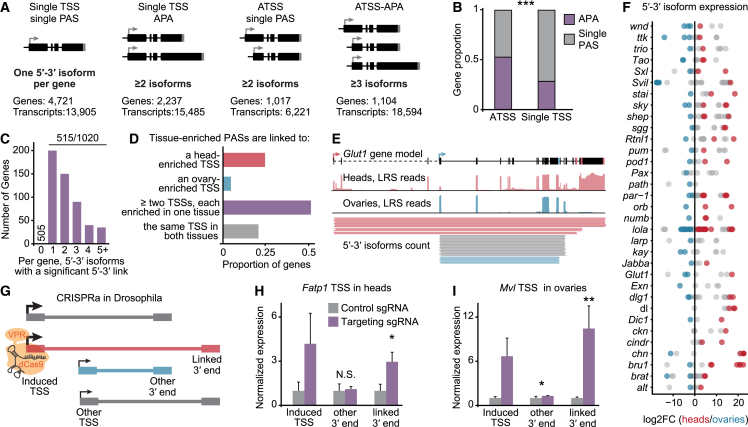
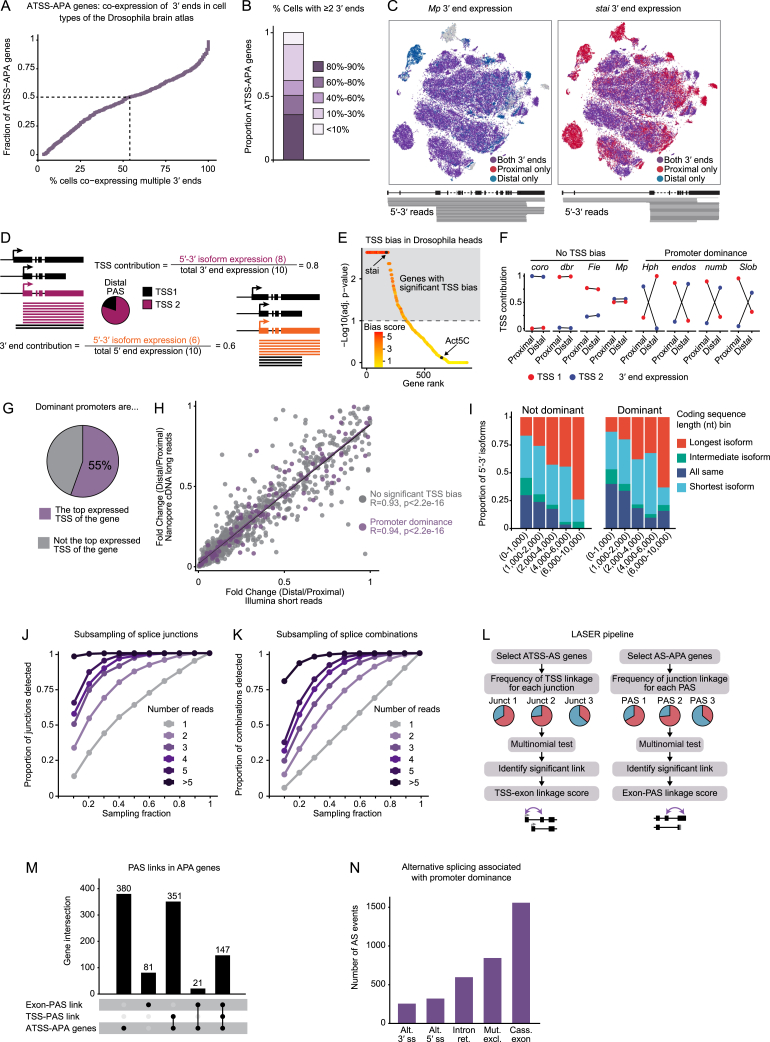
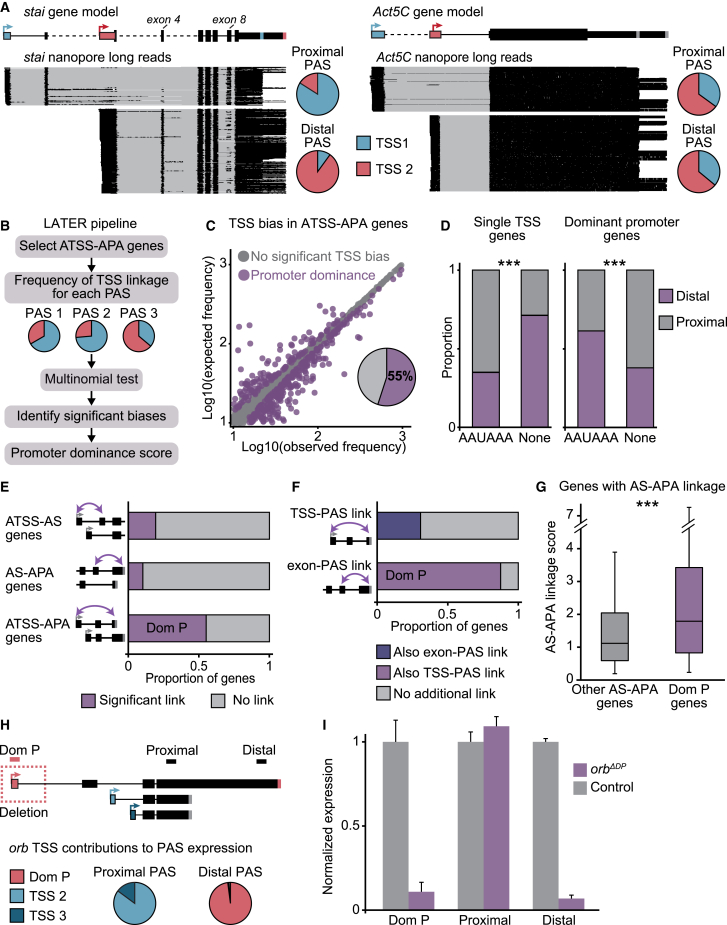
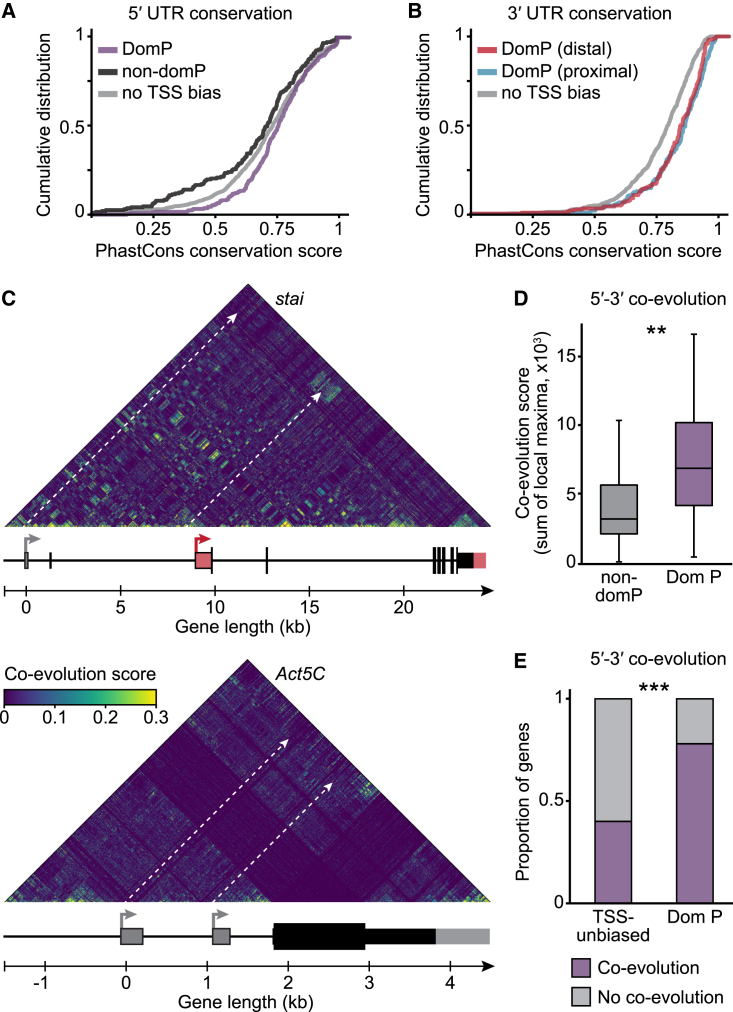
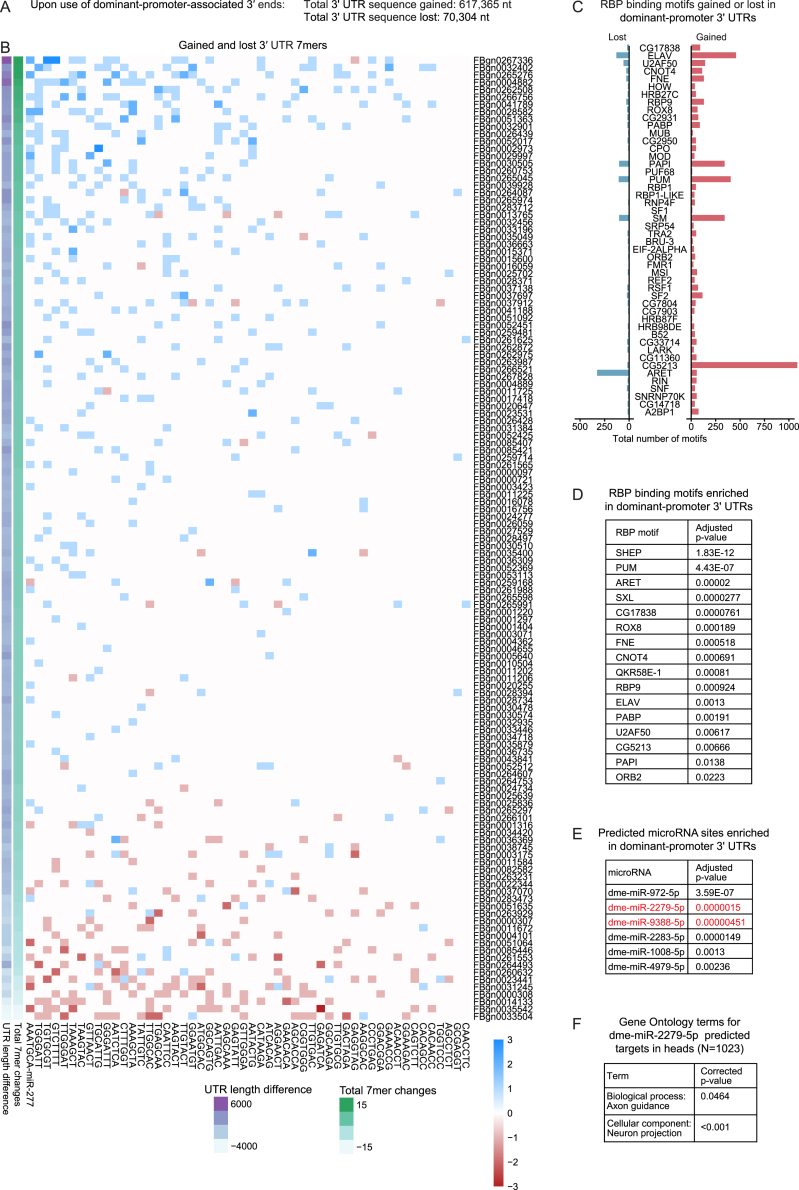
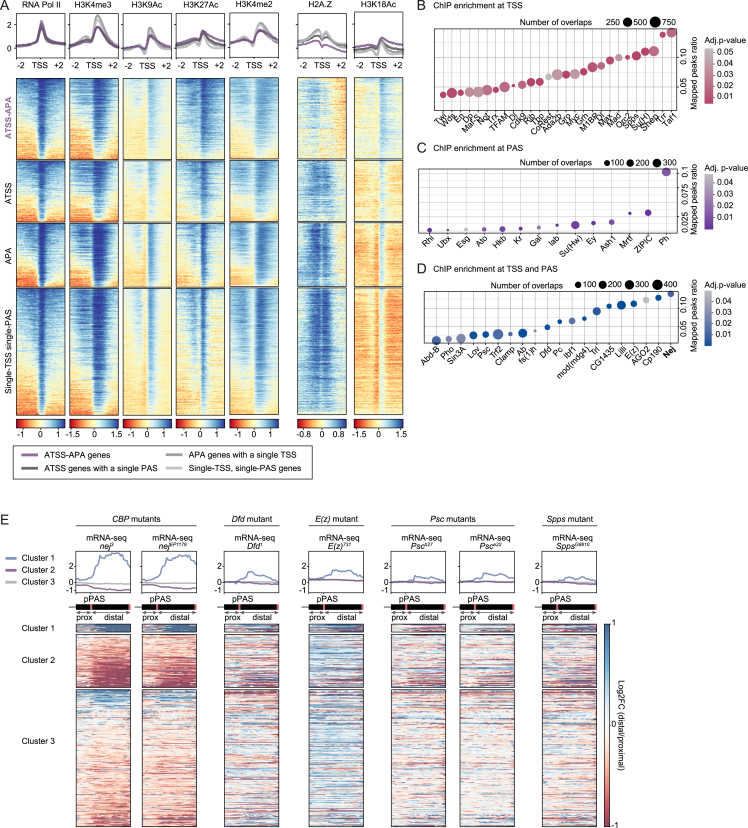
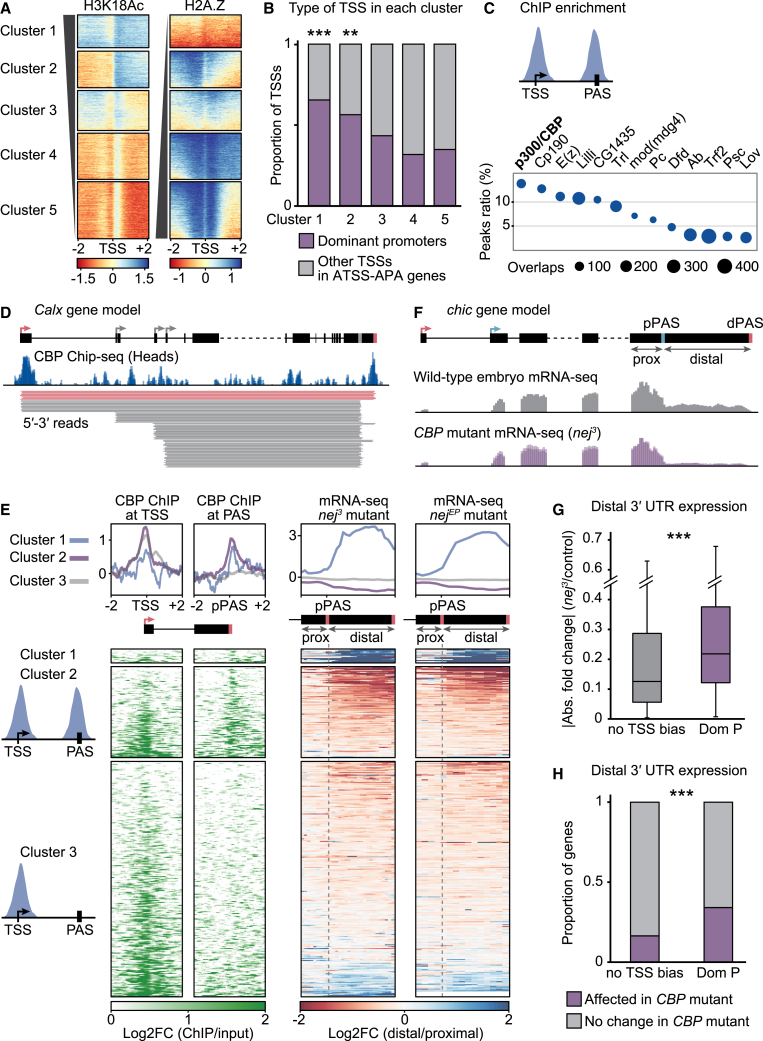
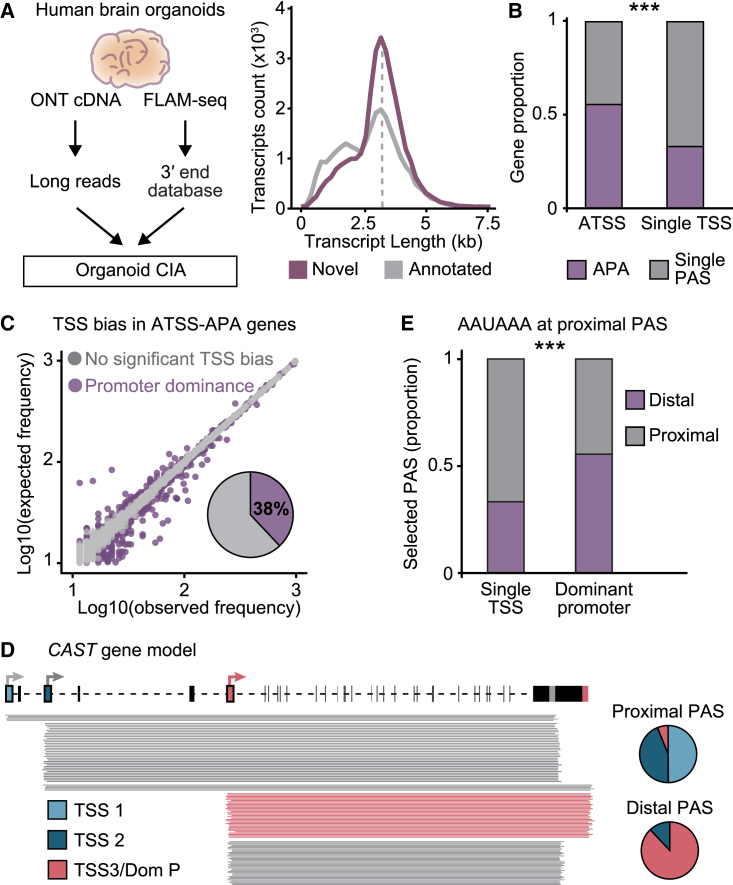
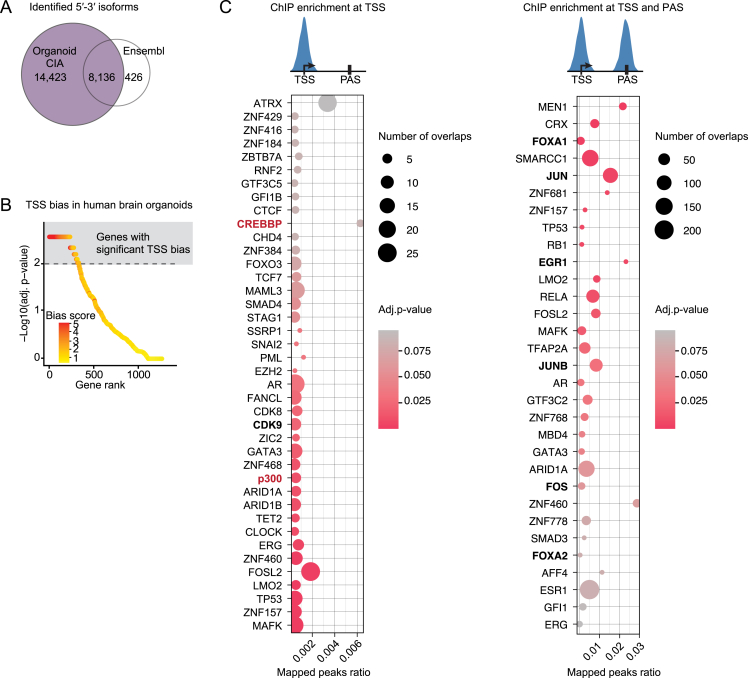
The generation of distinct messenger RNA isoforms through alternative RNA processing modulates the expression and function of genes, often in a cell-type-specific manner. Here, we assess the regulatory relationships between transcription initiation, alternative splicing, and 3' end site selection. Applying long-read sequencing to accurately represent even the longest transcripts from end to end, we quantify mRNA isoforms in Drosophila tissues, including the transcriptionally complex nervous system. We find that in Drosophila heads, as well as in human cerebral organoids, 3' end site choice is globally influenced by the site of transcription initiation (TSS). "Dominant promoters," characterized by specific epigenetic signatures including p300/CBP binding, impose a transcriptional constraint to define splice and polyadenylation variants. In vivo deletion or overexpression of dominant promoters as well as p300/CBP loss disrupted the 3' end expression landscape. Our study demonstrates the crucial impact of TSS choice on the regulation of transcript diversity and tissue identity.
Keywords: 5ʹ-3ʹ coupling; Drosophila; alternative polyadenylation; human brain organoids; long-read sequencing; mRNA isoform; nervous system; p300/CBP; transcription; transcription start site.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Comment in
-
The promoter as a trip navigator: Guiding alternative polyadenylation site destinations.Mol Cell. 2023 Jul 20;83(14):2395-2397. doi: 10.1016/j.molcel.2023.06.022. Mol Cell. 2023. PMID: 37478824
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
