

Animal models and mechanisms of tobacco smoke-induced chronic obstructive pulmonary disease (COPD)
- PMID: 37183431
- PMCID: PMC10718174
- DOI: 10.1080/10937404.2023.2208886
Animal models and mechanisms of tobacco smoke-induced chronic obstructive pulmonary disease (COPD)
Abstract
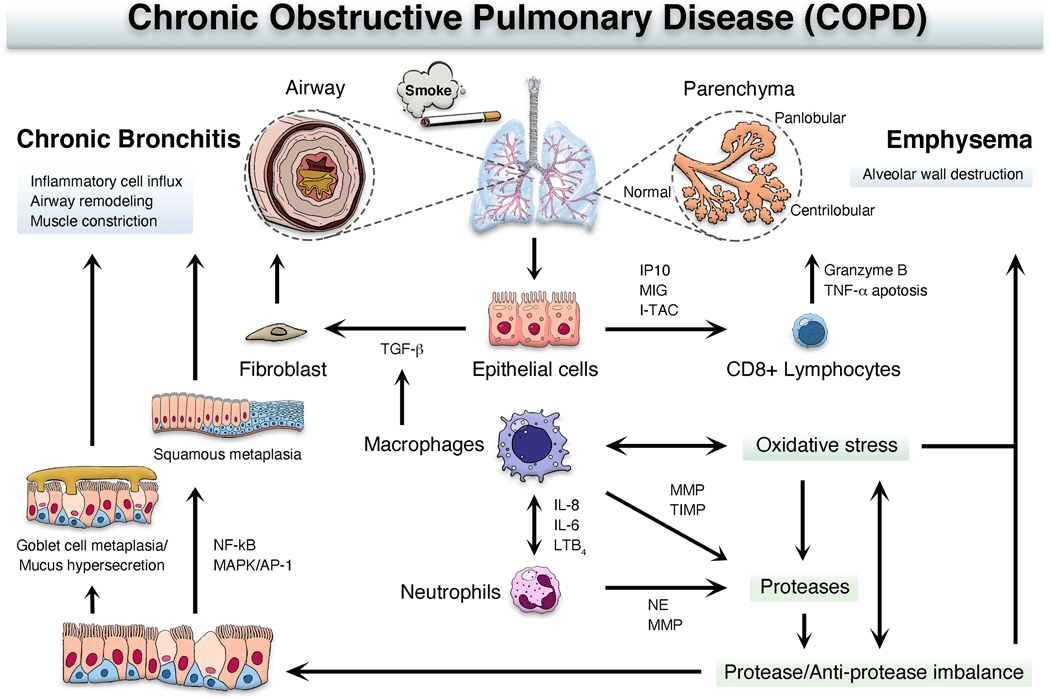
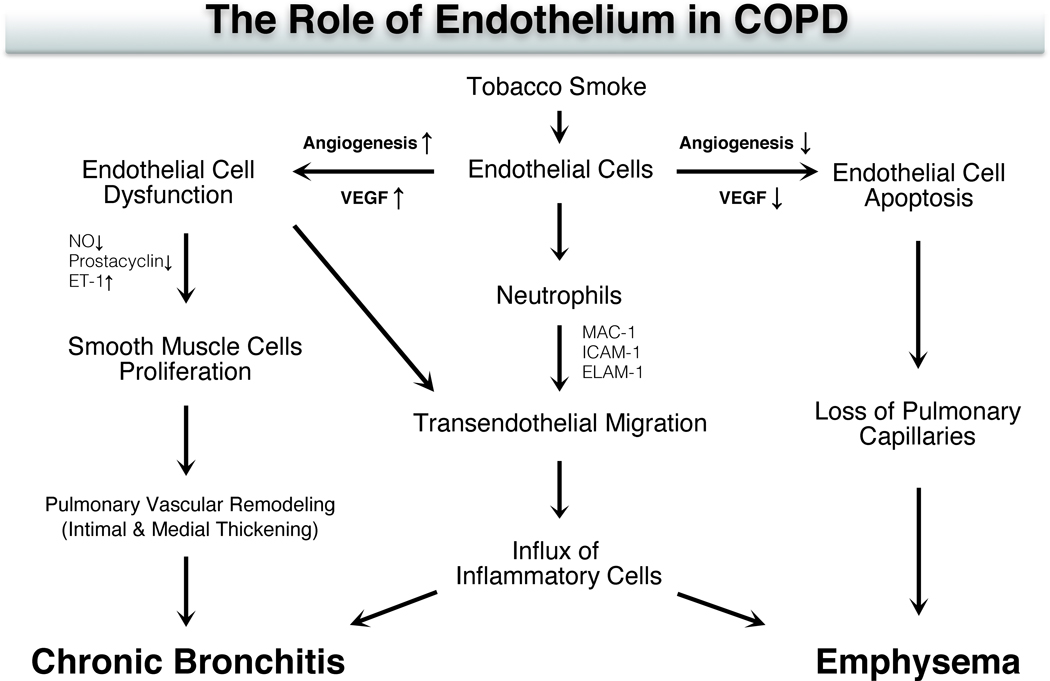
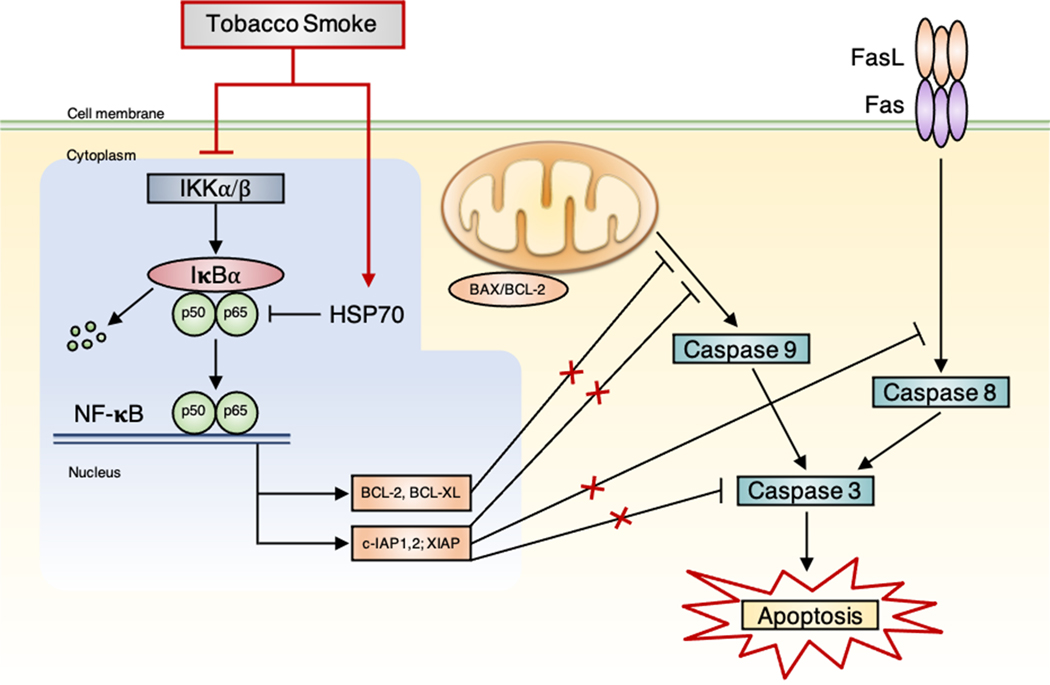
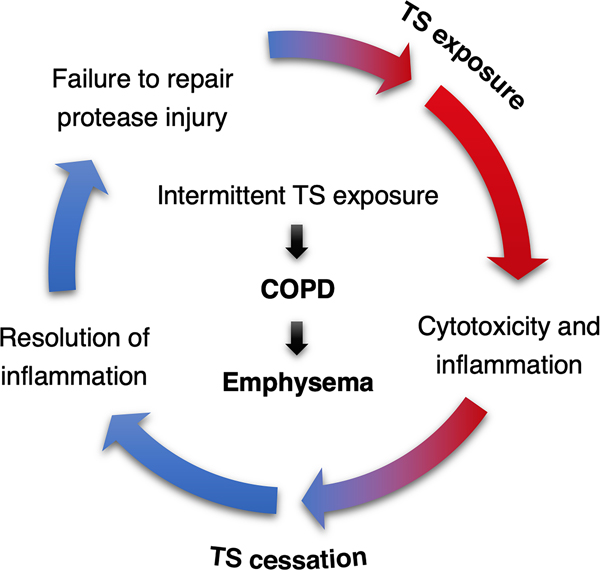
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide, and its global health burden is increasing. COPD is characterized by emphysema, mucus hypersecretion, and persistent lung inflammation, and clinically by chronic airflow obstruction and symptoms of dyspnea, cough, and fatigue in patients. A cluster of pathologies including chronic bronchitis, emphysema, asthma, and cardiovascular disease in the form of hypertension and atherosclerosis variably coexist in COPD patients. Underlying causes for COPD include primarily tobacco use but may also be driven by exposure to air pollutants, biomass burning, and workplace related fumes and chemicals. While no single animal model might mimic all features of human COPD, a wide variety of published models have collectively helped to improve our understanding of disease processes involved in the genesis and persistence of COPD. In this review, the pathogenesis and associated risk factors of COPD are examined in different mammalian models of the disease. Each animal model included in this review is exclusively created by tobacco smoke (TS) exposure. As animal models continue to aid in defining the pathobiological mechanisms of and possible novel therapeutic interventions for COPD, the advantages and disadvantages of each animal model are discussed.
Keywords: animal models; chronic bronchitis; chronic obstructive pulmonary disease (COPD); emphysema; tobacco smoke.
Figures
References
-
- Agusti A, Calverley PM, Celli B, Coxson HO, Edwards LD, Lomas DA, MacNee W, Miller BE, Rennard S, Silverman EK, Tal-Singer R, Wouters E, Yates JC, Vestbo J, and COPD longitudinally to identify predictive surrogate endpoints investigators evaluation of 2010. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res 11:122. - PubMed
-
- Andersen ZJ, Hvidberg M, Jensen SS, Ketzel M, Loft S, Sorensen M, Tjonneland A, Overvad K, and Raaschou-Nielsen O. 2011. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution: A cohort study. Am J Respir Crit Care Med 183: 455–461. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials