

Teasing out missing reactions in genome-scale metabolic networks through hypergraph learning
- PMID: 37185345
- PMCID: PMC10130184
- DOI: 10.1038/s41467-023-38110-7
Teasing out missing reactions in genome-scale metabolic networks through hypergraph learning
Abstract
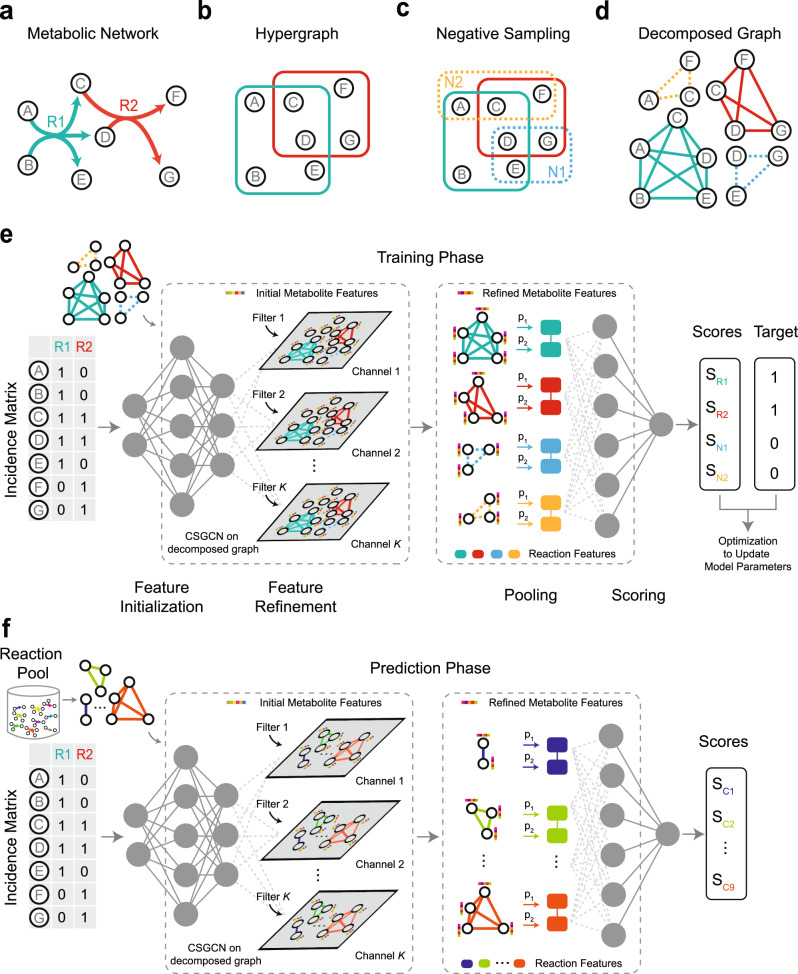
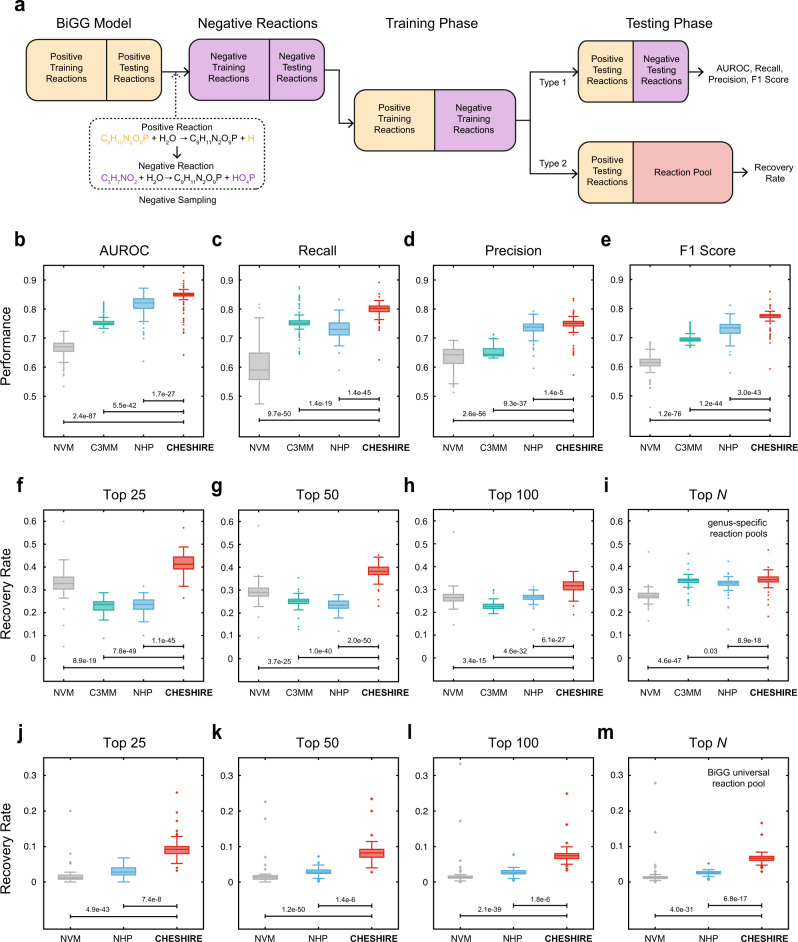
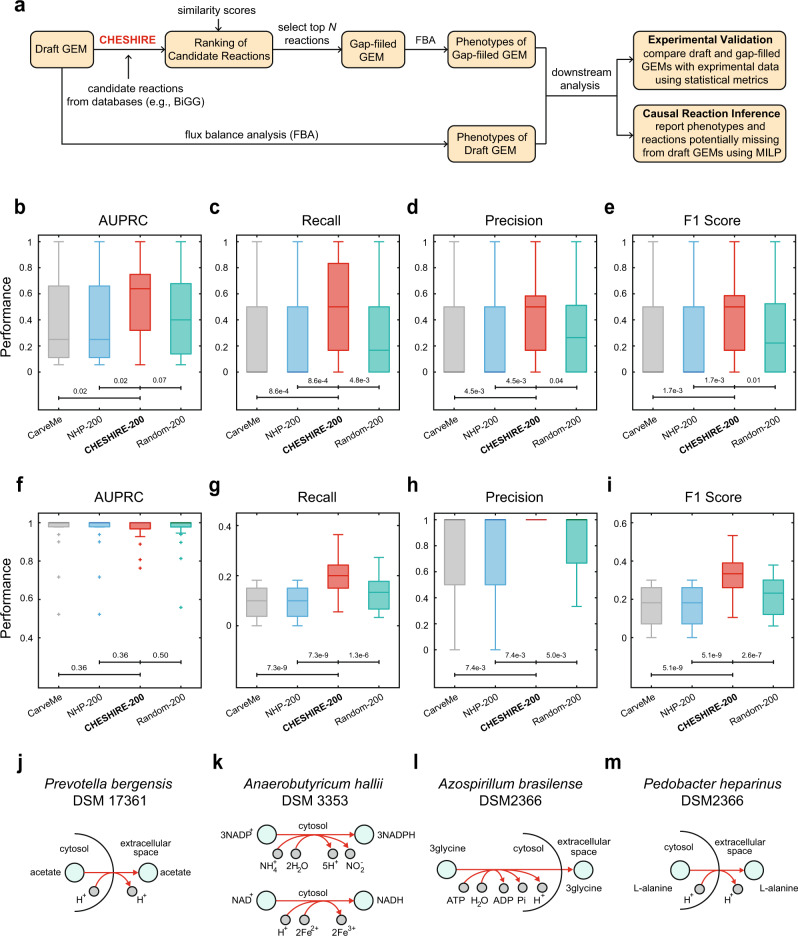
GEnome-scale Metabolic models (GEMs) are powerful tools to predict cellular metabolism and physiological states in living organisms. However, due to our imperfect knowledge of metabolic processes, even highly curated GEMs have knowledge gaps (e.g., missing reactions). Existing gap-filling methods typically require phenotypic data as input to tease out missing reactions. We still lack a computational method for rapid and accurate gap-filling of metabolic networks before experimental data is available. Here we present a deep learning-based method - CHEbyshev Spectral HyperlInk pREdictor (CHESHIRE) - to predict missing reactions in GEMs purely from metabolic network topology. We demonstrate that CHESHIRE outperforms other topology-based methods in predicting artificially removed reactions over 926 high- and intermediate-quality GEMs. Furthermore, CHESHIRE is able to improve the phenotypic predictions of 49 draft GEMs for fermentation products and amino acids secretions. Both types of validation suggest that CHESHIRE is a powerful tool for GEM curation to reveal unknown links between reactions and observed metabolic phenotypes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
