

Mechanistic roles of metal- and ligand-protonated species in hydrogen evolution with [Cp*Rh] complexes
- PMID: 37186841
- PMCID: PMC10214172
- DOI: 10.1073/pnas.2217189120
Mechanistic roles of metal- and ligand-protonated species in hydrogen evolution with [Cp*Rh] complexes
Abstract
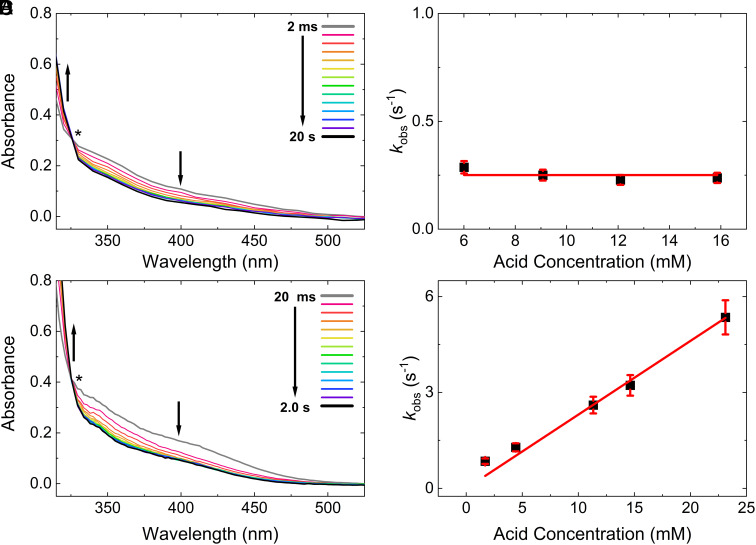
Protonation reactions involving organometallic complexes are ubiquitous in redox chemistry and often result in the generation of reactive metal hydrides. However, some organometallic species supported by η5-pentamethylcyclopentadienyl (Cp*) ligands have recently been shown to undergo ligand-centered protonation by direct proton transfer from acids or tautomerization of metal hydrides, resulting in the generation of complexes bearing the uncommon η4-pentamethylcyclopentadiene (Cp*H) ligand. Here, time-resolved pulse radiolysis (PR) and stopped-flow spectroscopic studies have been applied to examine the kinetics and atomistic details involved in the elementary electron- and proton-transfer steps leading to complexes ligated by Cp*H, using Cp*Rh(bpy) as a molecular model (where bpy is 2,2'-bipyridyl). Stopped-flow measurements coupled with infrared and UV-visible detection reveal that the sole product of initial protonation of Cp*Rh(bpy) is [Cp*Rh(H)(bpy)]+, an elusive hydride complex that has been spectroscopically and kinetically characterized here. Tautomerization of the hydride leads to the clean formation of [(Cp*H)Rh(bpy)]+. Variable-temperature and isotopic labeling experiments further confirm this assignment, providing experimental activation parameters and mechanistic insight into metal-mediated hydride-to-proton tautomerism. Spectroscopic monitoring of the second proton transfer event reveals that both the hydride and related Cp*H complex can be involved in further reactivity, showing that [(Cp*H)Rh] is not necessarily an off-cycle intermediate, but, instead, depending on the strength of the acid used to drive catalysis, an active participant in hydrogen evolution. Identification of the mechanistic roles of the protonated intermediates in the catalysis studied here could inform design of optimized catalytic systems supported by noninnocent cyclopentadienyl-type ligands.
Keywords: catalysis; energy; pulse radiolysis; redox chemistry; stopped-flow.
Conflict of interest statement
The authors declare no competing interest.
Figures





References
-
- Wilkinson G., Rosenblum M., Whiting M. C., Woodward R. B., The structure of iron bis-cyclopentadienyl. J. Am. Chem. Soc. 74, 2125–2126 (1952).
-
- Piper T. S., Wilkinson G., Alkyl and aryl derivatives of π-cyclopentadienyl compounds of chromium, molybdenum, tungsten, and iron. J. Inorg. Nucl. Chem. 3, 104–124 (1956).
-
- Lauher J. W., Hoffmann R., Structure and chemistry of bis(cyclopentadienyl)-MLn complexes. J. Am. Chem. Soc. 98, 1729–1742 (1976).
-
- Kuwana T., Bublitz D. E., Hoh G., Chronopotentiometric studies on the oxidation of ferrocene, ruthenocene, osmocene and some of their derivatives. J. Am. Chem. Soc. 82, 5811–5817 (1960).
-
- Pedersen A., Tilset M., Solvent-induced reductive elimination of pentamethylcyclopentadiene from a (pentamethylcyclopentadienyl)metal hydride. Organometallics 12, 3064–3068 (1993).
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
