

Functional characterization of C21ORF2 association with the NEK1 kinase mutated in human in diseases
- PMID: 37188479
- PMCID: PMC10185812
- DOI: 10.26508/lsa.202201740
Functional characterization of C21ORF2 association with the NEK1 kinase mutated in human in diseases
Abstract
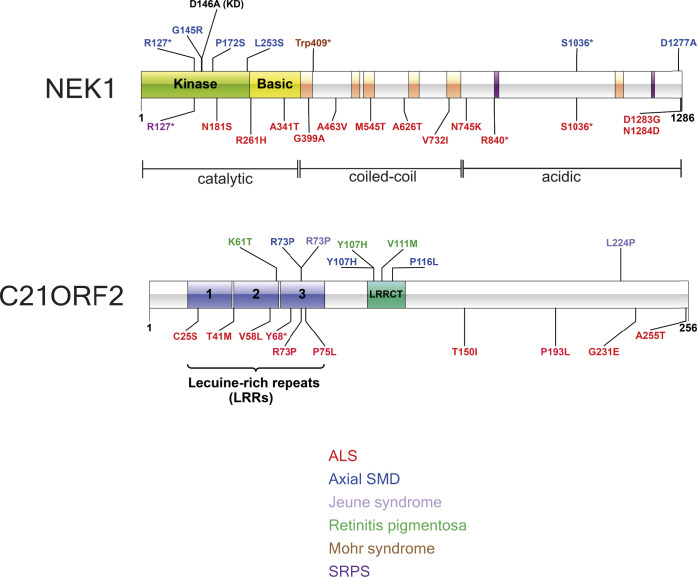
The NEK1 kinase controls ciliogenesis, mitosis, and DNA repair, and NEK1 mutations cause human diseases including axial spondylometaphyseal dysplasia and amyotrophic lateral sclerosis. C21ORF2 mutations cause a similar pattern of human diseases, suggesting close functional links with NEK1 Here, we report that endogenous NEK1 and C21ORF2 form a tight complex in human cells. A C21ORF2 interaction domain "CID" at the C-terminus of NEK1 is necessary for its association with C21ORF2 in cells, and pathogenic mutations in this region disrupt the complex. AlphaFold modelling predicts an extended binding interface between a leucine-rich repeat domain in C21ORF2 and the NEK1-CID, and our model may explain why pathogenic mutations perturb the complex. We show that NEK1 mutations that inhibit kinase activity or weaken its association with C21ORF2 severely compromise ciliogenesis, and that C21ORF2, like NEK1 is required for homologous recombination. These data enhance our understanding of how the NEK1 kinase is regulated, and they shed light on NEK1-C21ORF2-associated diseases.
© 2023 Gregorczyk et al.
Conflict of interest statement
D Durocher is a shareholder and advisor of Repare Therapeutics.
Figures













References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials