

Juvenile idiopathic inflammatory myositis: an update on pathophysiology and clinical care
- PMID: 37188756
- PMCID: PMC10184643
- DOI: 10.1038/s41584-023-00967-9
Juvenile idiopathic inflammatory myositis: an update on pathophysiology and clinical care
Abstract
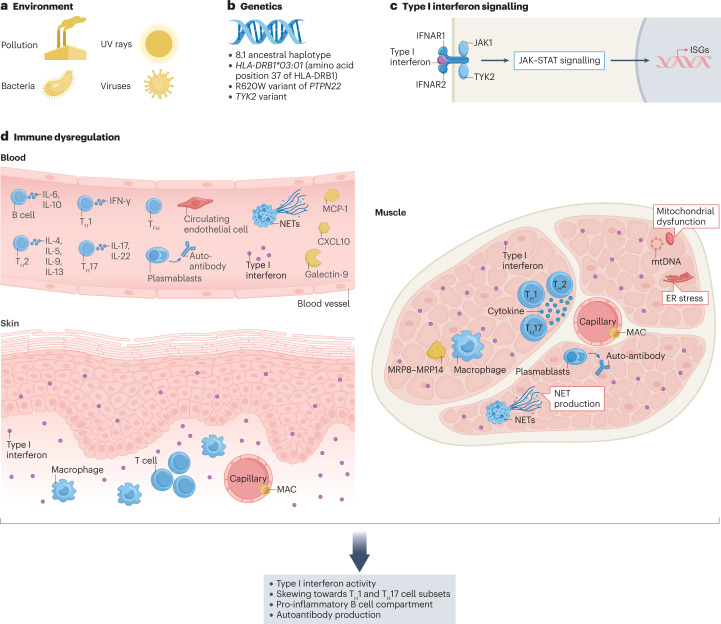
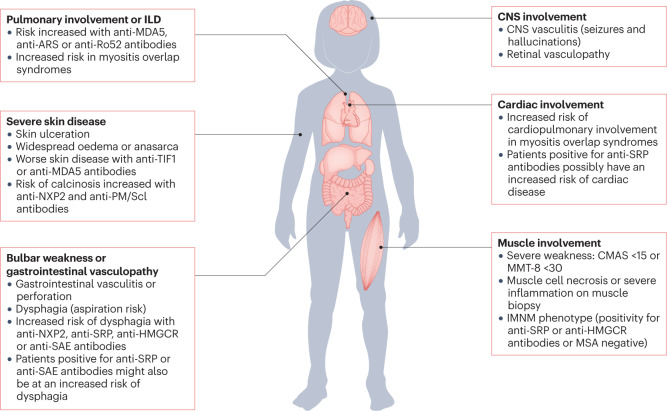
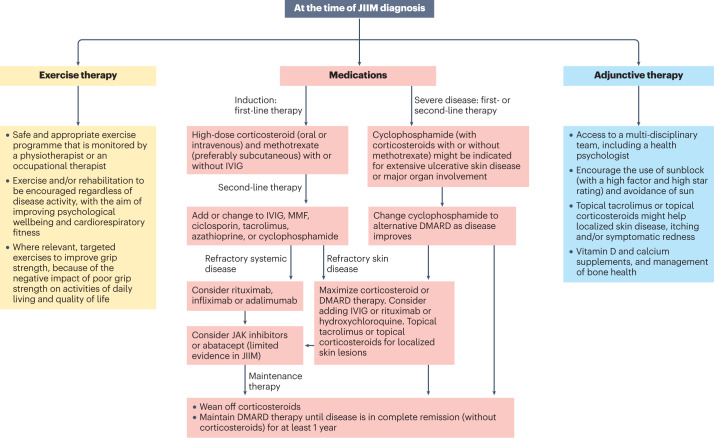
The childhood-onset or juvenile idiopathic inflammatory myopathies (JIIMs) are a heterogenous group of rare and serious autoimmune diseases of children and young people that predominantly affect the muscles and skin but can also involve other organs, including the lungs, gut, joints, heart and central nervous system. Different myositis-specific autoantibodies have been identified that are associated with different muscle biopsy features, as well as with different clinical characteristics, prognoses and treatment responses. Thus, myositis-specific autoantibodies can be used to subset JIIMs into sub-phenotypes; some of these sub-phenotypes parallel disease seen in adults, whereas others are distinct from adult-onset idiopathic inflammatory myopathies. Although treatments and management have much improved over the past decade, evidence is still lacking for many of the current treatments and few validated prognostic biomarkers are available with which to predict response to treatment, comorbidities (such as calcinosis) or outcome. Emerging data on the pathogenesis of the JIIMs are leading to proposals for new trials and tools for monitoring disease.
© 2023. Springer Nature Limited.
Conflict of interest statement
All authors declare that the UK JDM Cohort and Biomarker Study (JDCBS) is currently funded by grants from the Great Ormond Street Hospital (GOSH) Children’s Charity, the National Institute for Health Research (NIHR) Biomedical Research Centre at GOSH, Cure JM, Myositis UK, Versus Arthritis and Remission Charity. L.R.W. declares that she is a consultant for Pfizer Inc (with consulting fees going wholly to the University College London). The other authors all declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
