

The ClusPro AbEMap web server for the prediction of antibody epitopes
- PMID: 37188806
- PMCID: PMC10898366
- DOI: 10.1038/s41596-023-00826-7
The ClusPro AbEMap web server for the prediction of antibody epitopes
Abstract
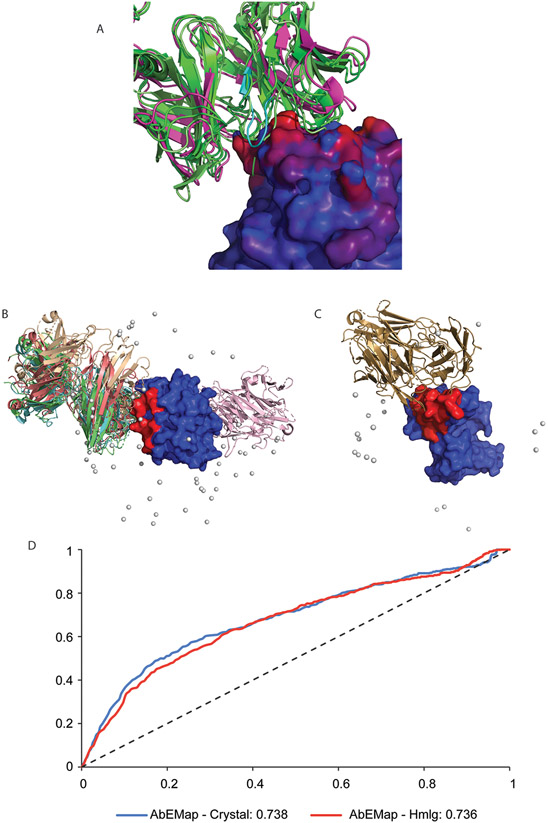
Antibodies play an important role in the immune system by binding to molecules called antigens at their respective epitopes. These interfaces or epitopes are structural entities determined by the interactions between an antibody and an antigen, making them ideal systems to analyze by using docking programs. Since the advent of high-throughput antibody sequencing, the ability to perform epitope mapping using only the sequence of the antibody has become a high priority. ClusPro, a leading protein-protein docking server, together with its template-based modeling version, ClusPro-TBM, have been re-purposed to map epitopes for specific antibody-antigen interactions by using the Antibody Epitope Mapping server (AbEMap). ClusPro-AbEMap offers three different modes for users depending on the information available on the antibody as follows: (i) X-ray structure, (ii) computational/predicted model of the structure or (iii) only the amino acid sequence. The AbEMap server presents a likelihood score for each antigen residue of being part of the epitope. We provide detailed information on the server's capabilities for the three options and discuss how to obtain the best results. In light of the recent introduction of AlphaFold2 (AF2), we also show how one of the modes allows users to use their AF2-generated antibody models as input. The protocol describes the relative advantages of the server compared to other epitope-mapping tools, its limitations and potential areas of improvement. The server may take 45-90 min depending on the size of the proteins.
© 2023. Springer Nature Limited.
Figures










References
-
- Desta IT et al. Proteins. (2022) DOI: 10.1002/prot.26420 - DOI
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
