

Systematic identification of anticancer drug targets reveals a nucleus-to-mitochondria ROS-sensing pathway
- PMID: 37192619
- PMCID: PMC10225361
- DOI: 10.1016/j.cell.2023.04.026
Systematic identification of anticancer drug targets reveals a nucleus-to-mitochondria ROS-sensing pathway
Abstract
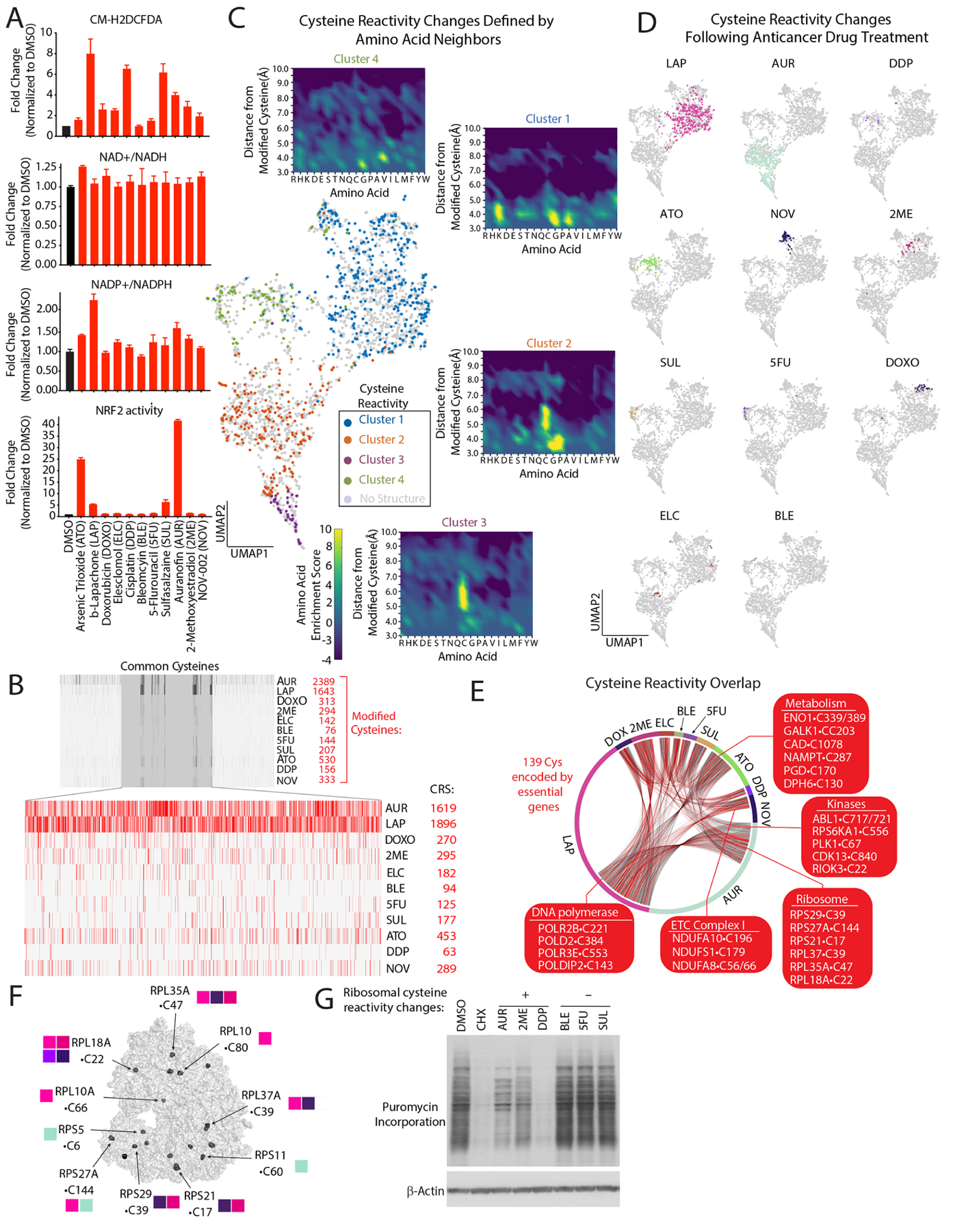
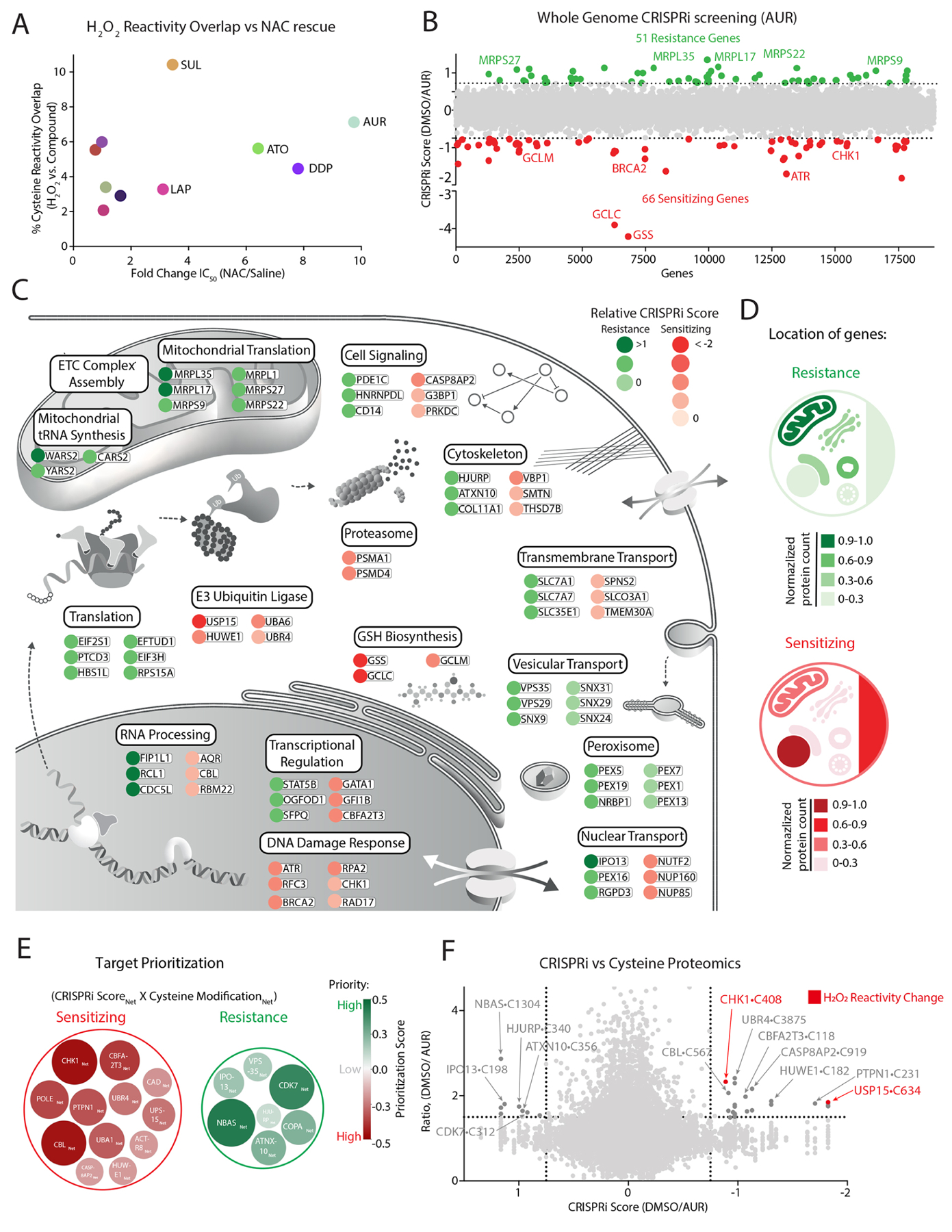
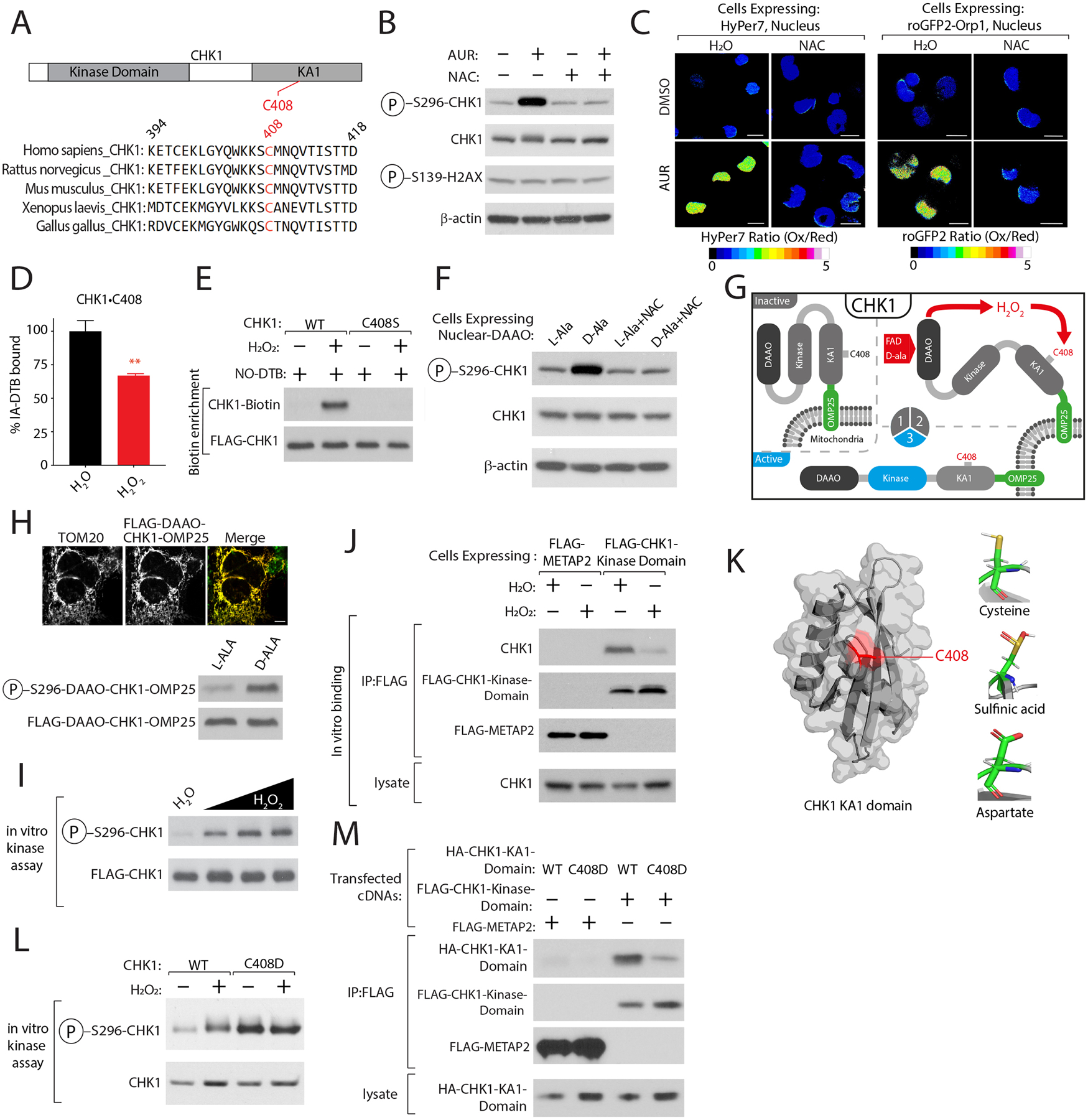
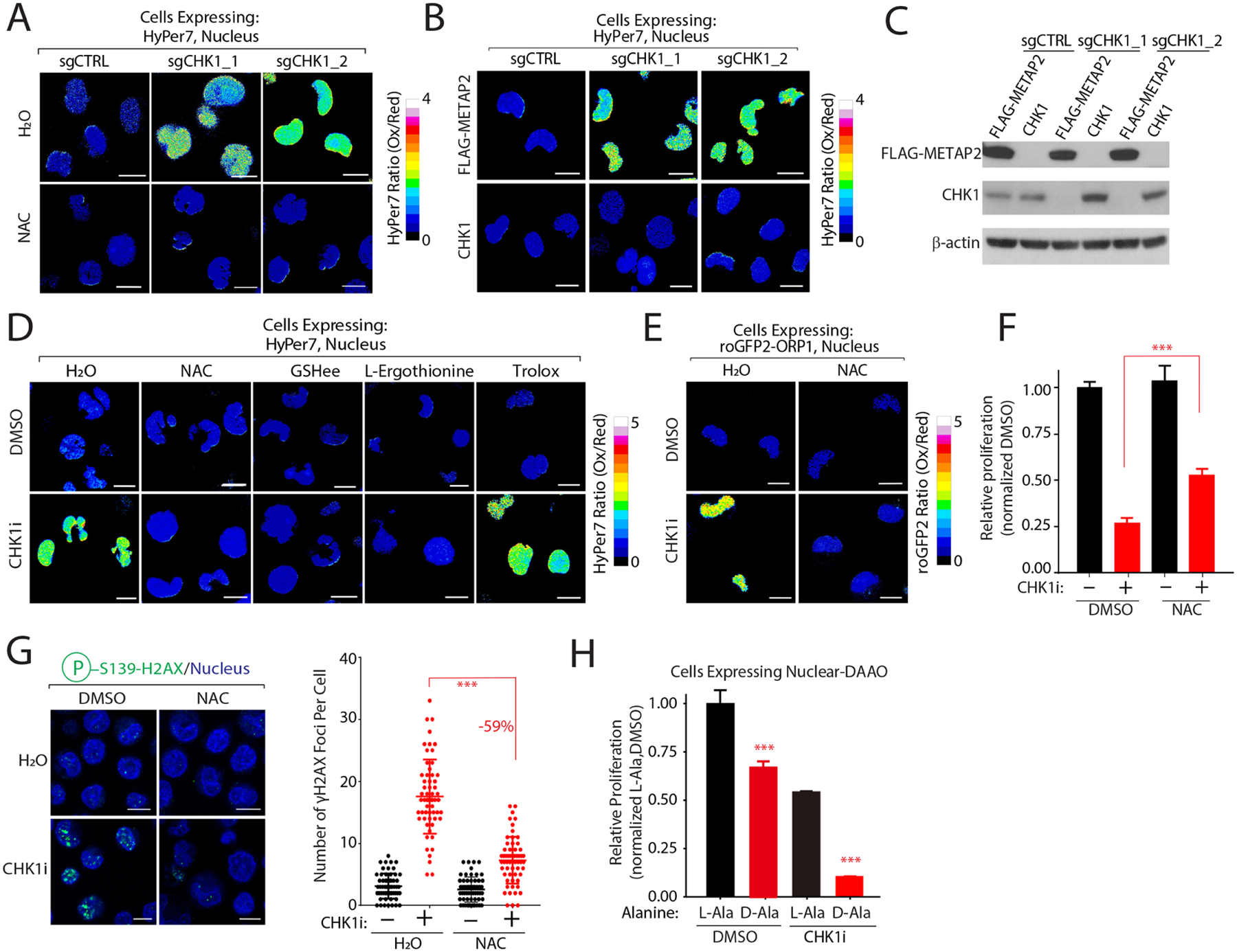
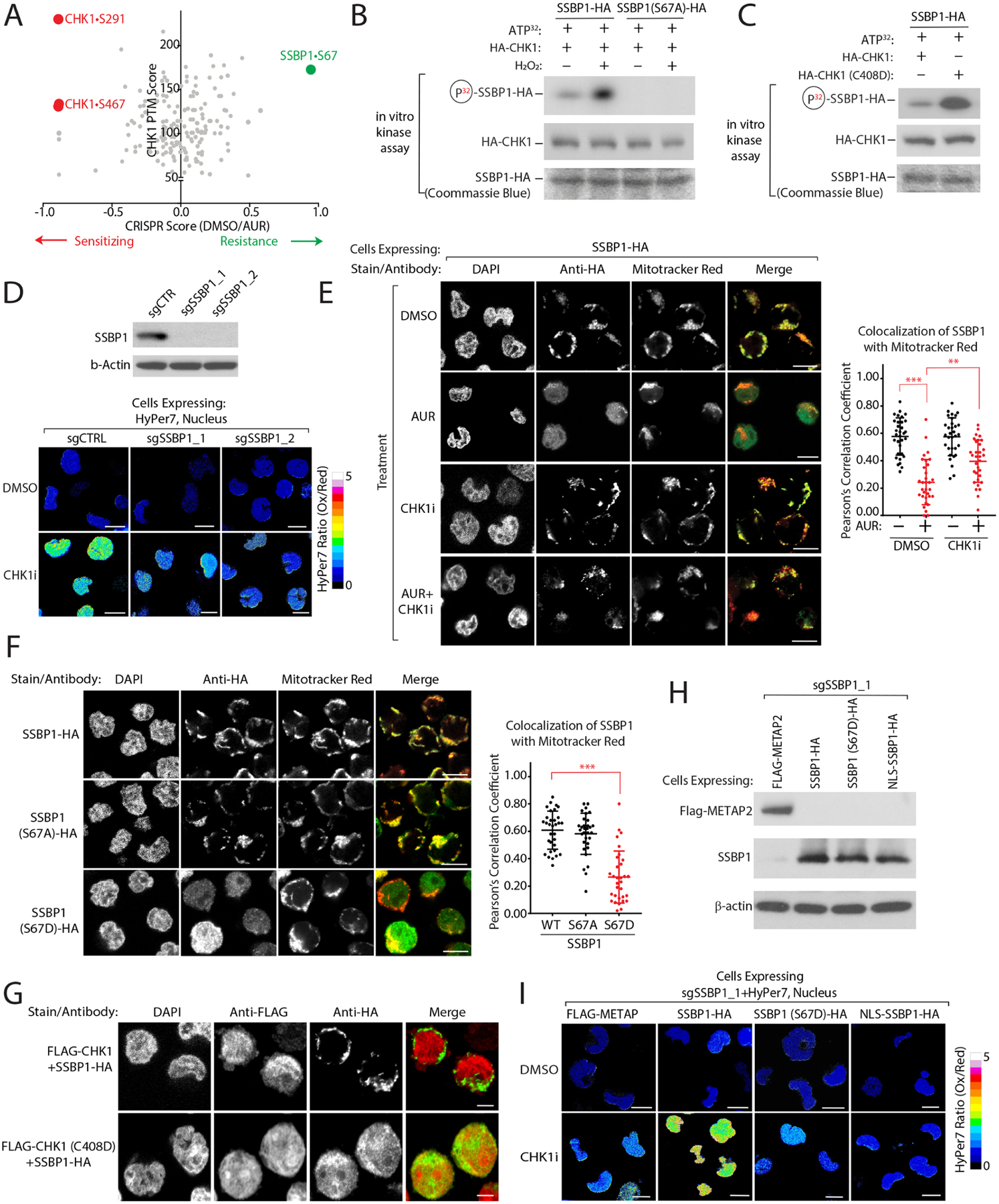
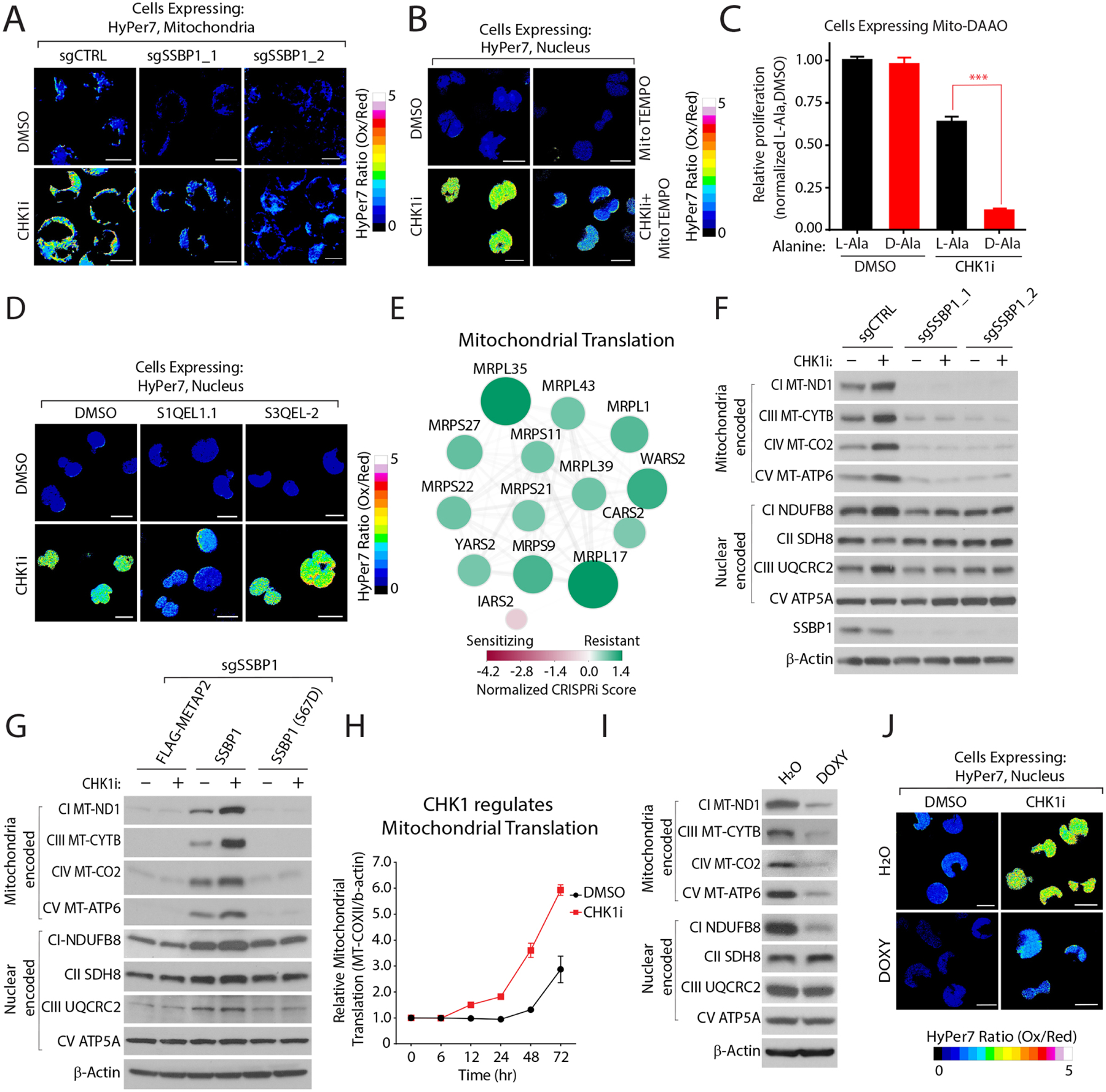
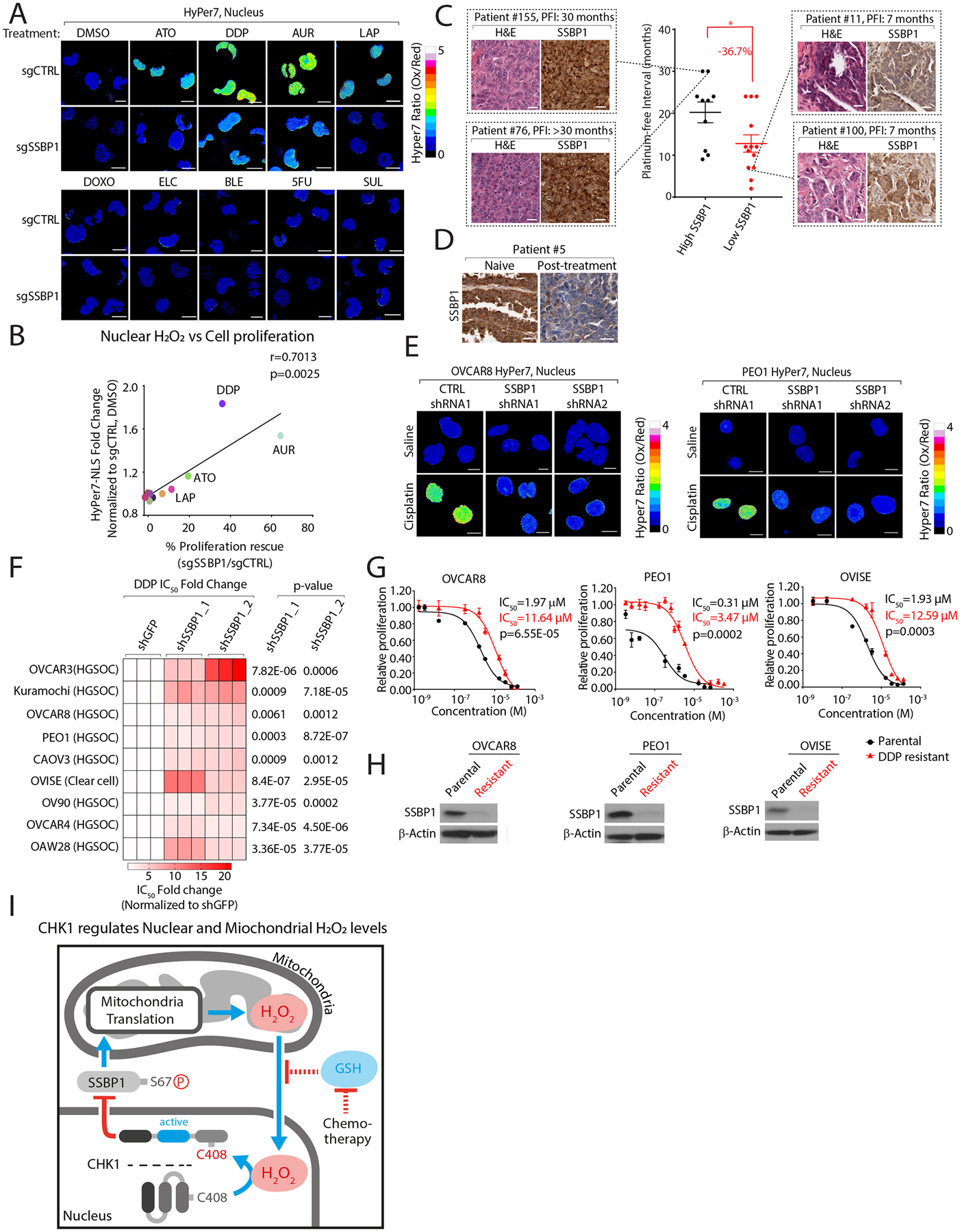
Multiple anticancer drugs have been proposed to cause cell death, in part, by increasing the steady-state levels of cellular reactive oxygen species (ROS). However, for most of these drugs, exactly how the resultant ROS function and are sensed is poorly understood. It remains unclear which proteins the ROS modify and their roles in drug sensitivity/resistance. To answer these questions, we examined 11 anticancer drugs with an integrated proteogenomic approach identifying not only many unique targets but also shared ones-including ribosomal components, suggesting common mechanisms by which drugs regulate translation. We focus on CHK1 that we find is a nuclear H2O2 sensor that launches a cellular program to dampen ROS. CHK1 phosphorylates the mitochondrial DNA-binding protein SSBP1 to prevent its mitochondrial localization, which in turn decreases nuclear H2O2. Our results reveal a druggable nucleus-to-mitochondria ROS-sensing pathway-required to resolve nuclear H2O2 accumulation and mediate resistance to platinum-based agents in ovarian cancers.
Keywords: CHK1; chemical proteomics; chemoresistance; mitochondrial translation; nuclear ROS; nucleus-to-mitochondria signaling.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.P.P., C.M.S., M.P.S., and S.A.B. are employees of Cell Signaling Technology, Inc. L.B.-P. is a founder, consultant, and holds privately held equity in Scorpion Therapeutics.
Figures
Update of
-
Identification of chemotherapy targets reveals a nucleus-to-mitochondria ROS sensing pathway.bioRxiv [Preprint]. 2023 Mar 11:2023.03.11.532189. doi: 10.1101/2023.03.11.532189. bioRxiv. 2023. Update in: Cell. 2023 May 25;186(11):2361-2379.e25. doi: 10.1016/j.cell.2023.04.026. PMID: 36945474 Free PMC article. Updated. Preprint.
Comment in
-
A ROS-mediated resistance pathway to anticancer drugs.Nat Rev Drug Discov. 2023 Jul;22(7):534. doi: 10.1038/d41573-023-00094-w. Nat Rev Drug Discov. 2023. PMID: 37277606 No abstract available.
-
How CHKing ROS signaling preserves genomic integrity.Cell Res. 2023 Nov;33(11):815-816. doi: 10.1038/s41422-023-00840-0. Cell Res. 2023. PMID: 37380808 Free PMC article. No abstract available.
-
Cys-regulation: oxidized CHK1 controls cross-compartment circuit of chemoresistance.Trends Cancer. 2023 Sep;9(9):700-702. doi: 10.1016/j.trecan.2023.07.005. Epub 2023 Aug 1. Trends Cancer. 2023. PMID: 37532649
References
-
- Dixon SJ, and Stockwell BR (2019). The Hallmarks of Ferroptosis. Annual Review of Cancer Biology 3, 35–54. 10.1146/annurev-cancerbio-030518-055844. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
