

Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants
- PMID: 37193706
- PMCID: PMC10187524
- DOI: 10.1038/s41467-023-38435-3
Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants
Abstract
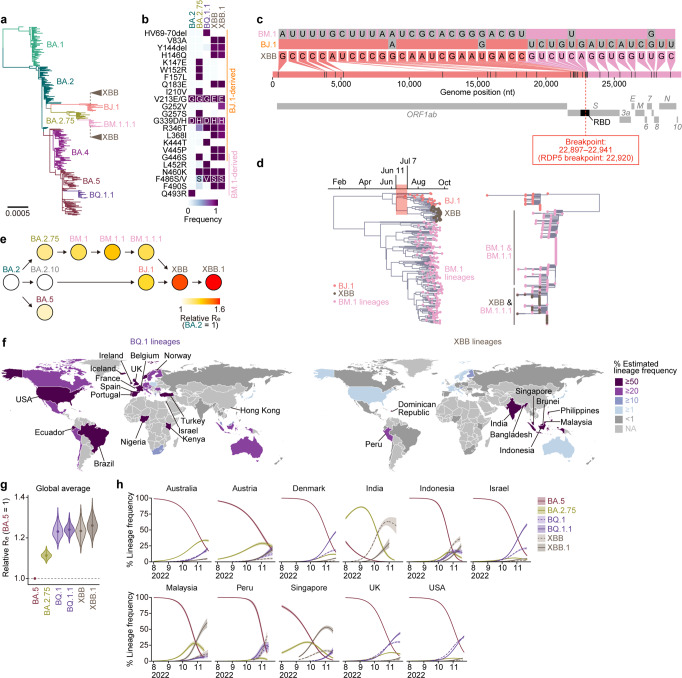
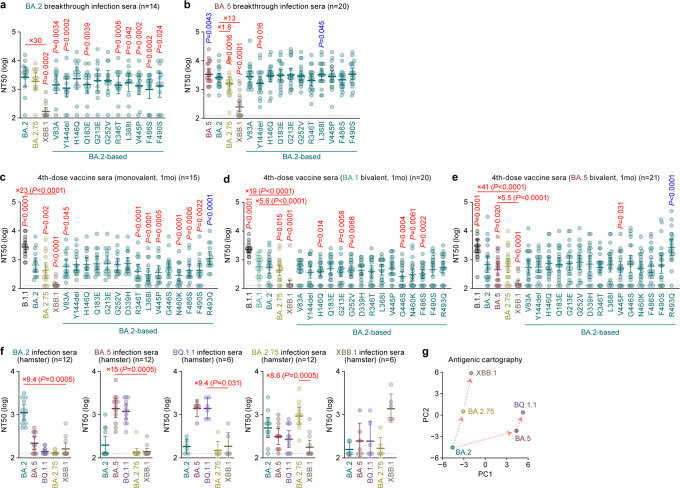
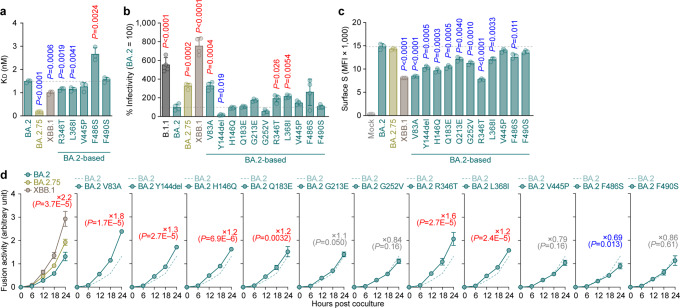
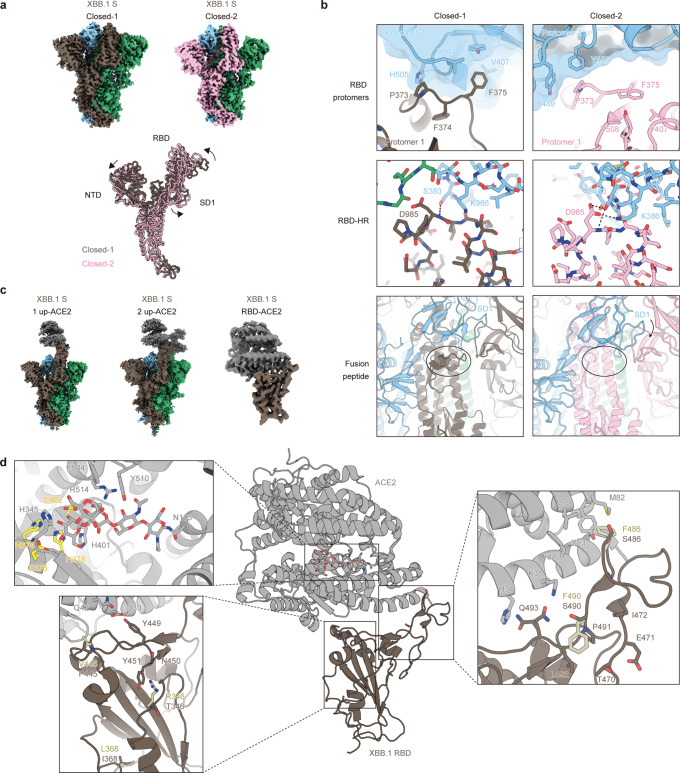
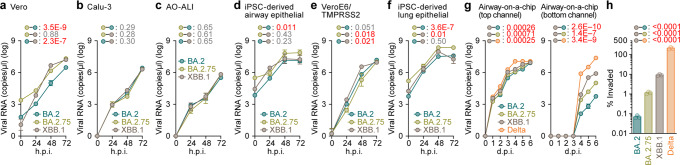
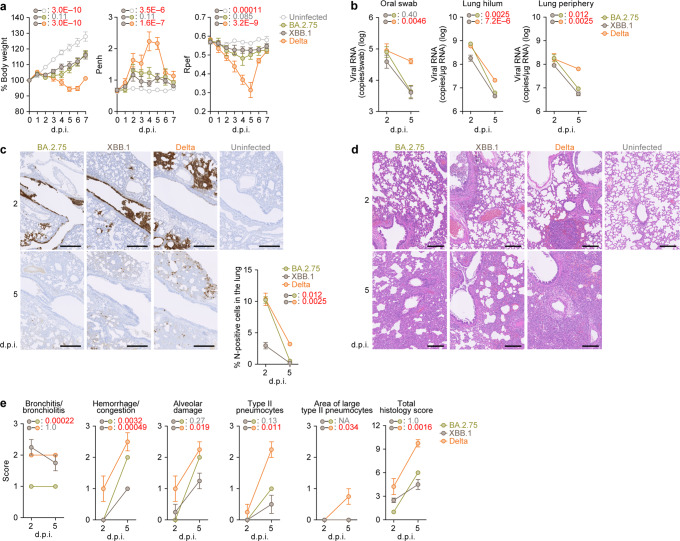
In late 2022, SARS-CoV-2 Omicron subvariants have become highly diversified, and XBB is spreading rapidly around the world. Our phylogenetic analyses suggested that XBB emerged through the recombination of two cocirculating BA.2 lineages, BJ.1 and BM.1.1.1 (a progeny of BA.2.75), during the summer of 2022. XBB.1 is the variant most profoundly resistant to BA.2/5 breakthrough infection sera to date and is more fusogenic than BA.2.75. The recombination breakpoint is located in the receptor-binding domain of spike, and each region of the recombinant spike confers immune evasion and increases fusogenicity. We further provide the structural basis for the interaction between XBB.1 spike and human ACE2. Finally, the intrinsic pathogenicity of XBB.1 in male hamsters is comparable to or even lower than that of BA.2.75. Our multiscale investigation provides evidence suggesting that XBB is the first observed SARS-CoV-2 variant to increase its fitness through recombination rather than substitutions.
© 2023. The Author(s).
Conflict of interest statement
Y.Y. and T.N. are founders and shareholders of HiLung, Inc. Y.Y. is a co-inventor of patents (PCT/JP2016/057254; “Method for inducing differentiation of alveolar epithelial cells”, PCT/JP2016/059786, “Method of producing airway epithelial cells”). The other authors declare that no competing interests exist.
Figures
References
-
- WHO. Tracking SARS-CoV-2 variants (March 30, 2023) https://www.who.int/en/activities/tracking-SARS-CoV-2-variants. (2022).
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
