

A weakly structured stem for human origins in Africa
- PMID: 37198480
- PMCID: PMC10208968
- DOI: 10.1038/s41586-023-06055-y
A weakly structured stem for human origins in Africa
Erratum in
-
Publisher Correction: A weakly structured stem for human origins in Africa.Nature. 2023 Aug;620(7972):E11. doi: 10.1038/s41586-023-06433-6. Nature. 2023. PMID: 37460744 Free PMC article. No abstract available.
Abstract
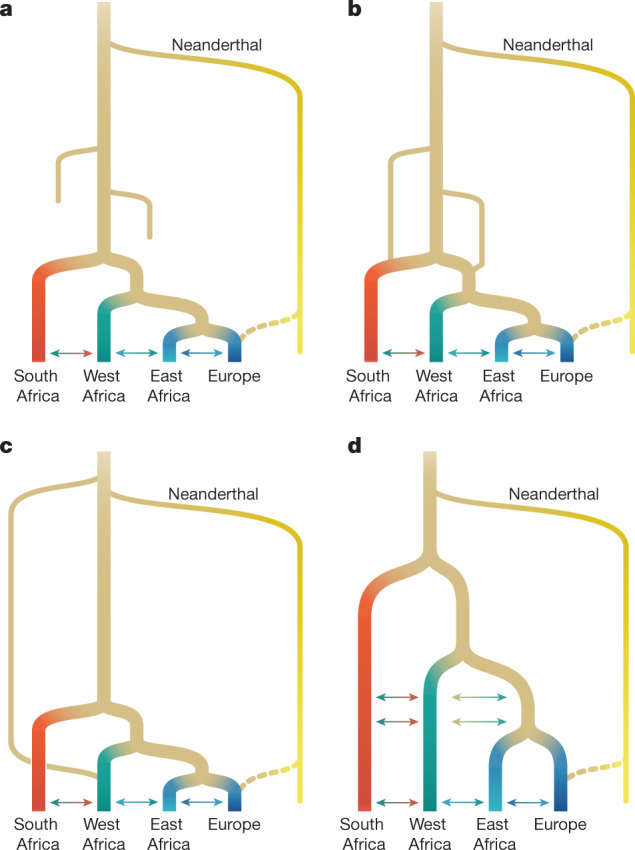
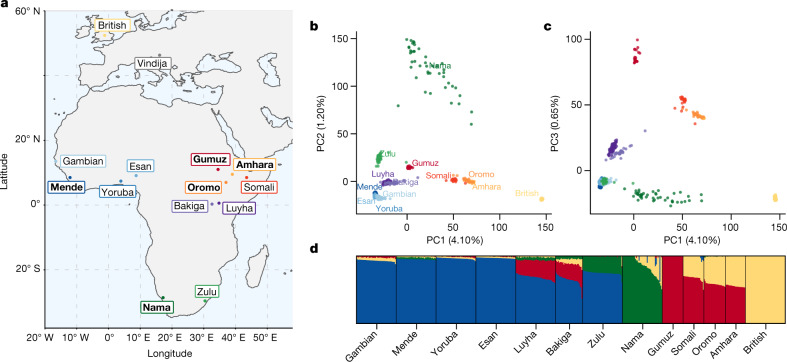
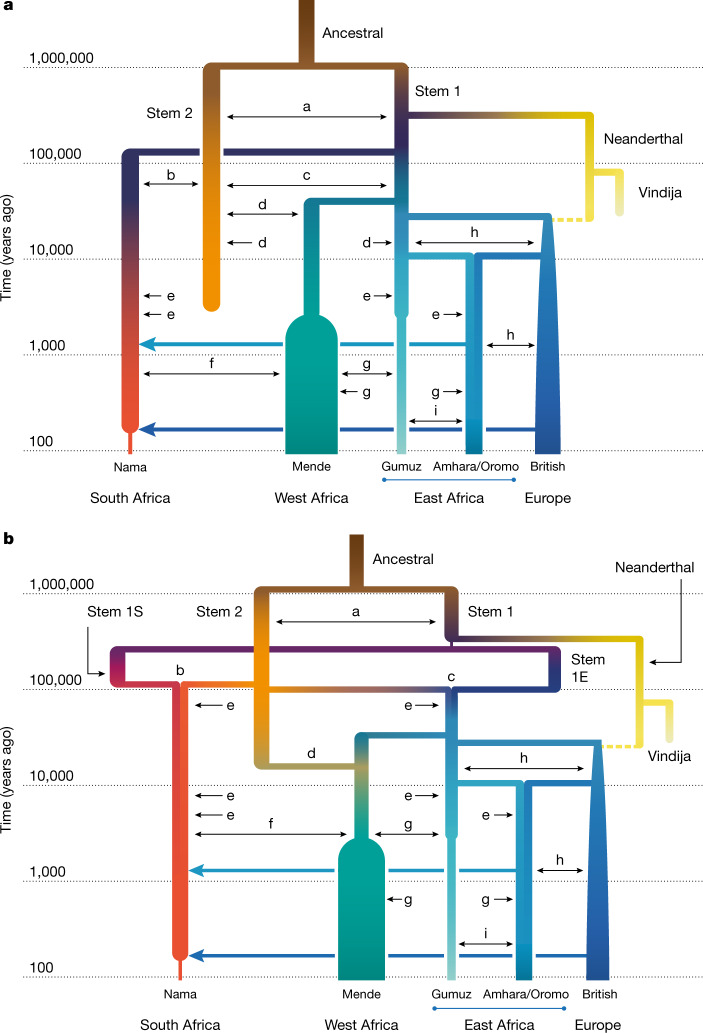
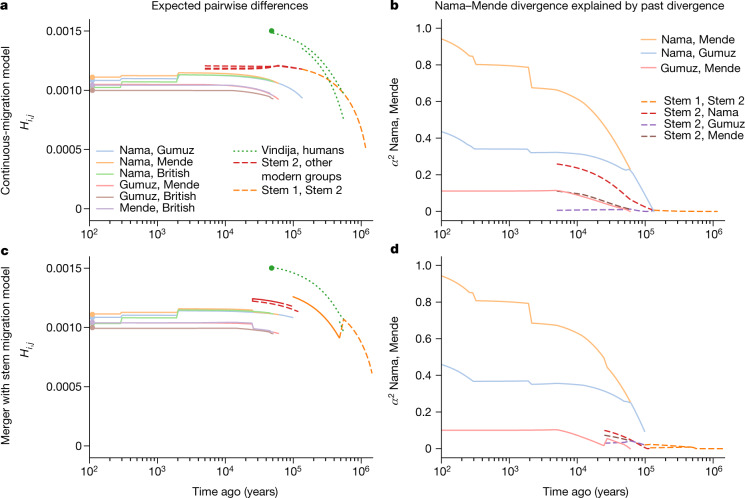
Despite broad agreement that Homo sapiens originated in Africa, considerable uncertainty surrounds specific models of divergence and migration across the continent1. Progress is hampered by a shortage of fossil and genomic data, as well as variability in previous estimates of divergence times1. Here we seek to discriminate among such models by considering linkage disequilibrium and diversity-based statistics, optimized for rapid, complex demographic inference2. We infer detailed demographic models for populations across Africa, including eastern and western representatives, and newly sequenced whole genomes from 44 Nama (Khoe-San) individuals from southern Africa. We infer a reticulated African population history in which present-day population structure dates back to Marine Isotope Stage 5. The earliest population divergence among contemporary populations occurred 120,000 to 135,000 years ago and was preceded by links between two or more weakly differentiated ancestral Homo populations connected by gene flow over hundreds of thousands of years. Such weakly structured stem models explain patterns of polymorphism that had previously been attributed to contributions from archaic hominins in Africa2-7. In contrast to models with archaic introgression, we predict that fossil remains from coexisting ancestral populations should be genetically and morphologically similar, and that only an inferred 1-4% of genetic differentiation among contemporary human populations can be attributed to genetic drift between stem populations. We show that model misspecification explains the variation in previous estimates of divergence times, and argue that studying a range of models is key to making robust inferences about deep history.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
One species, many roots?Nat Ecol Evol. 2023 Jul;7(7):975-976. doi: 10.1038/s41559-023-02080-2. Nat Ecol Evol. 2023. PMID: 37198291 No abstract available.
