

ERα-associated translocations underlie oncogene amplifications in breast cancer
- PMID: 37198482
- PMCID: PMC10307628
- DOI: 10.1038/s41586-023-06057-w
ERα-associated translocations underlie oncogene amplifications in breast cancer
Abstract
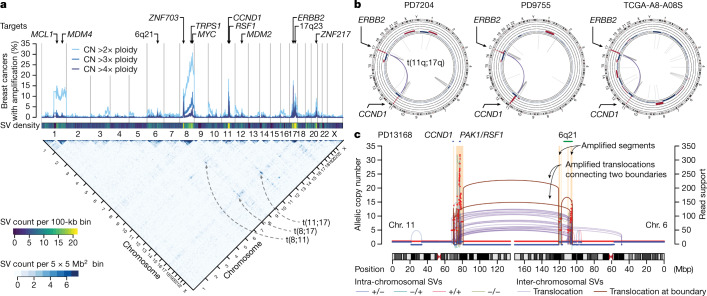
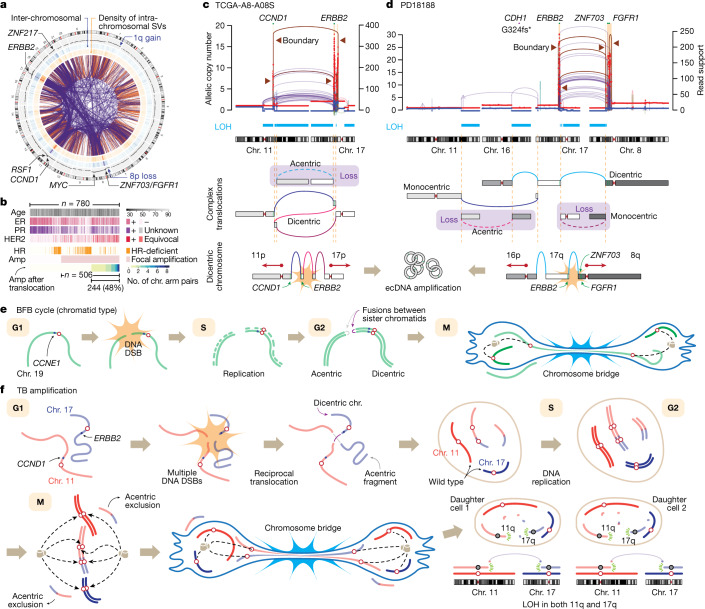
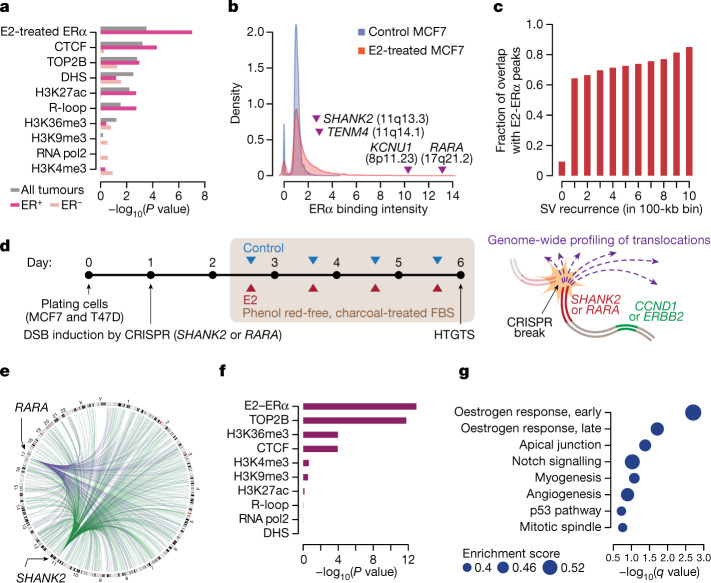
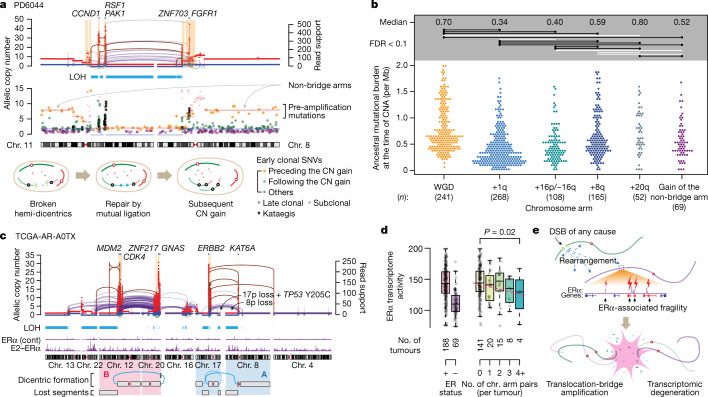
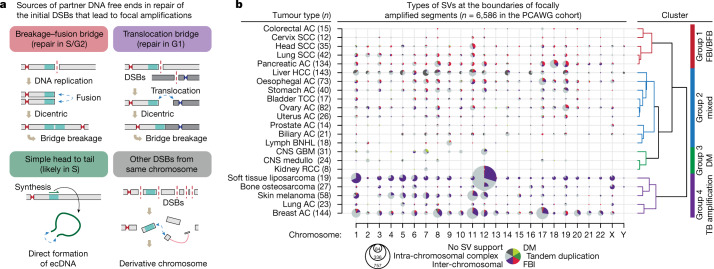
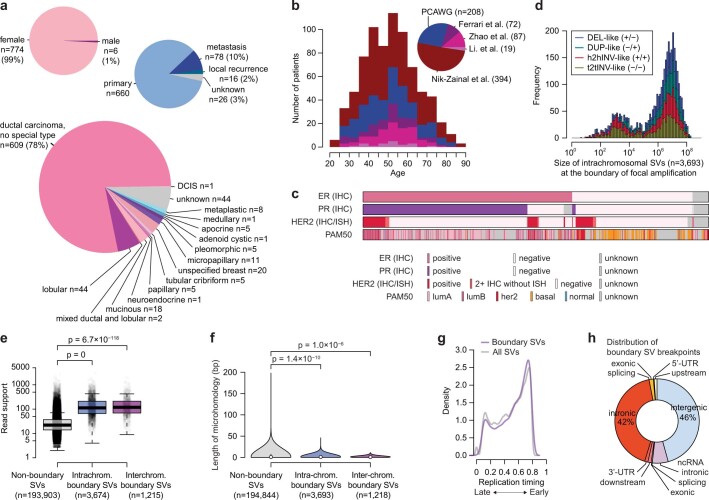
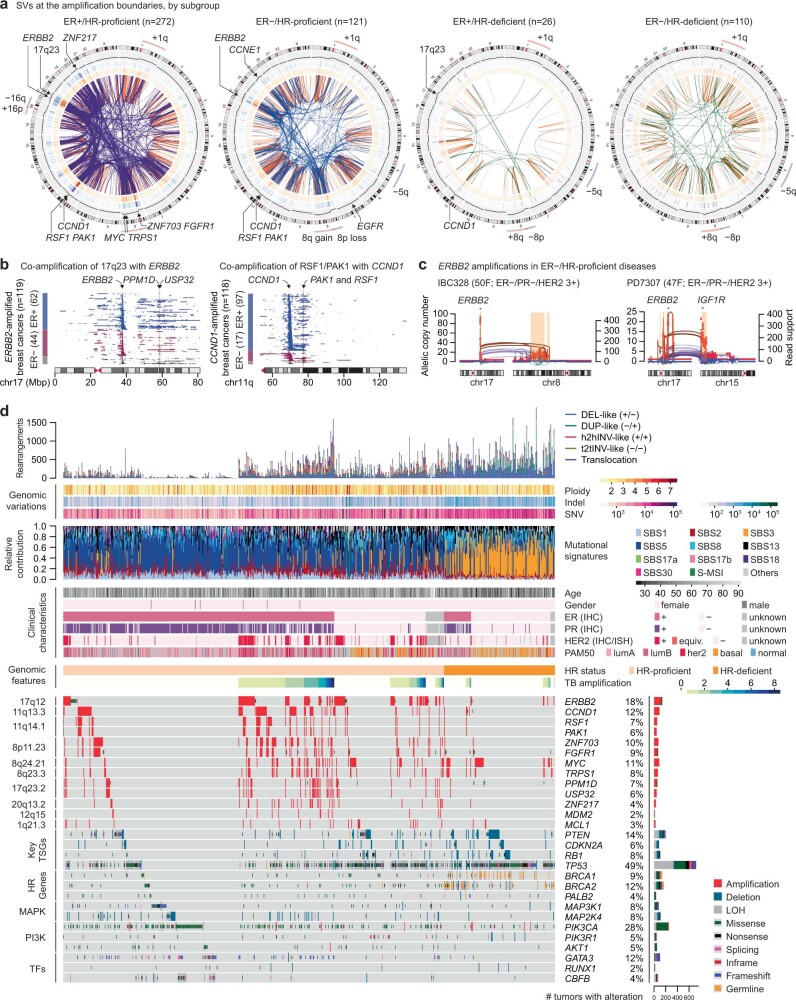
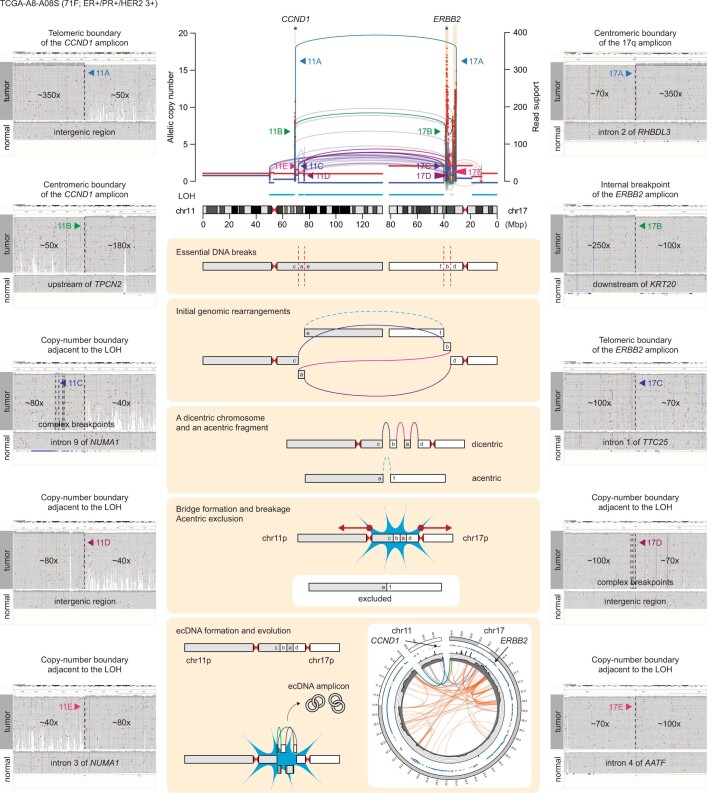
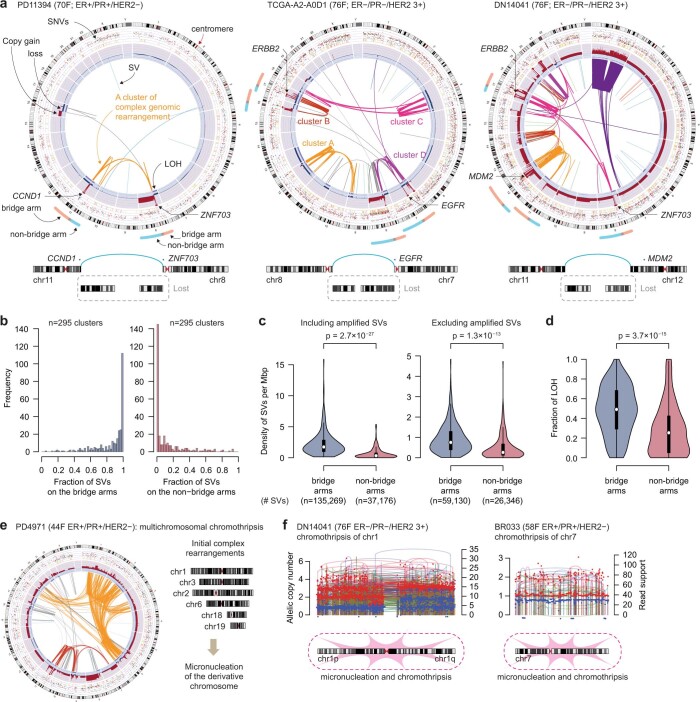
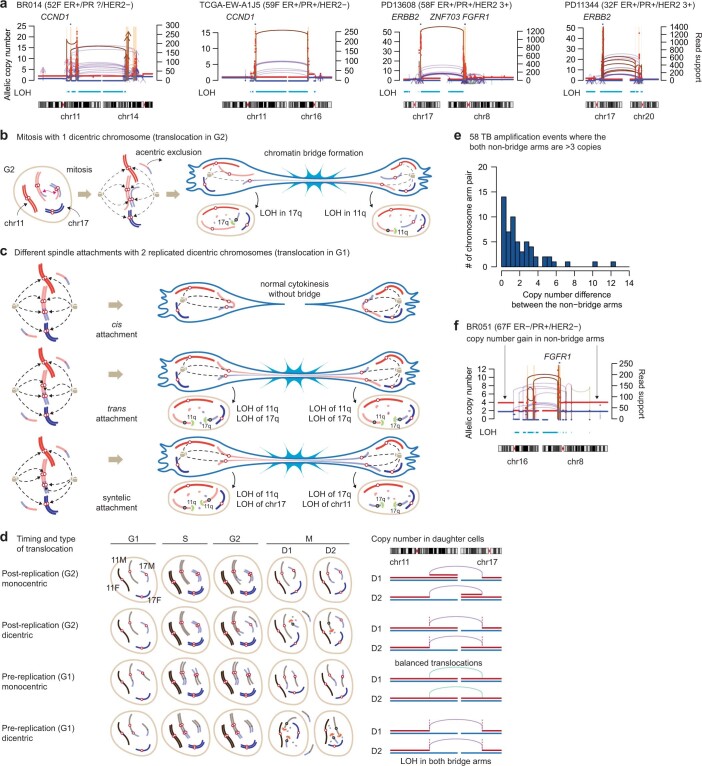
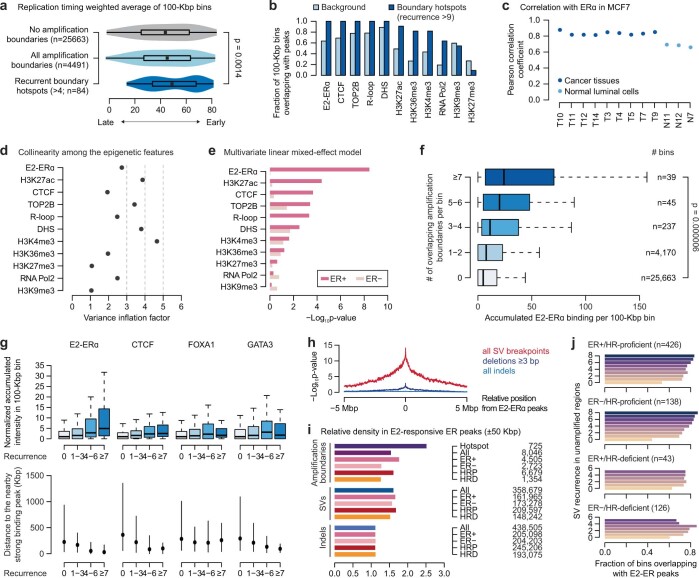
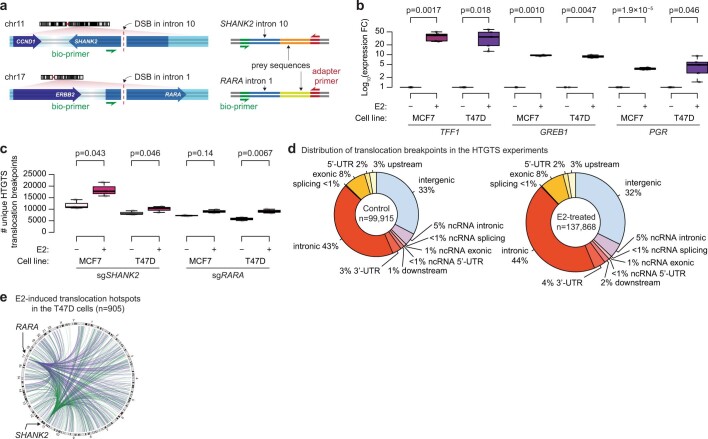
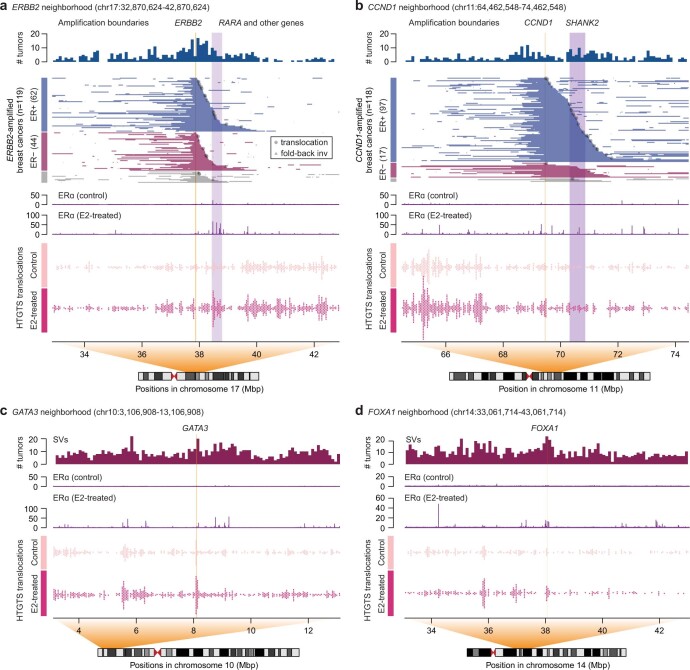
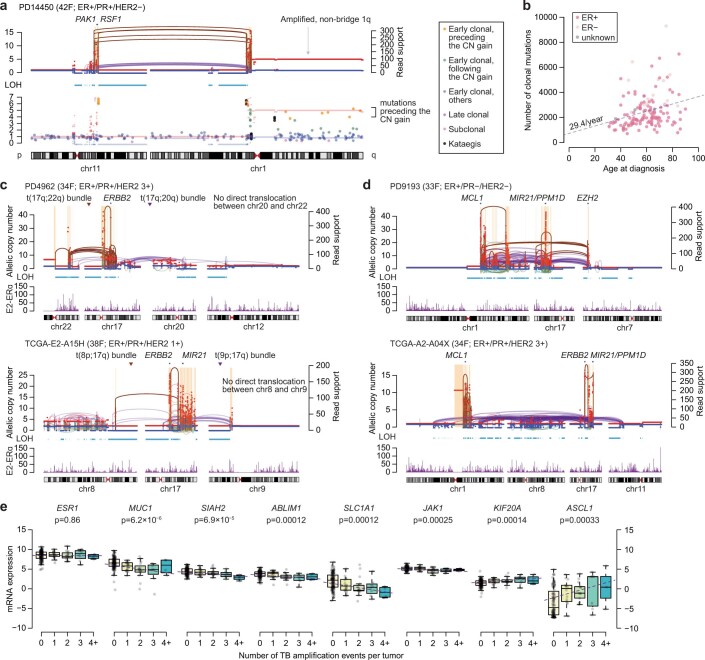
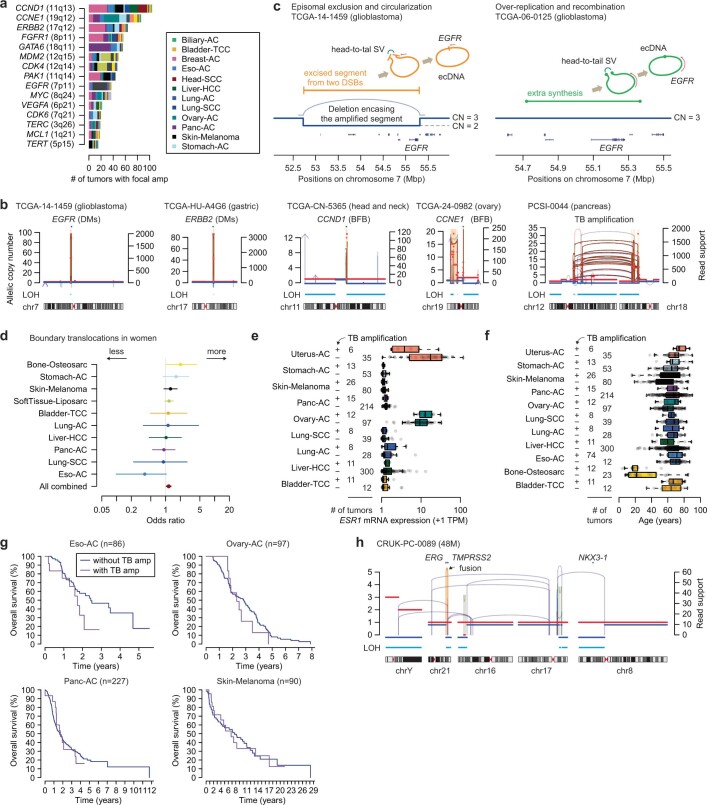
Focal copy-number amplification is an oncogenic event. Although recent studies have revealed the complex structure1-3 and the evolutionary trajectories4 of oncogene amplicons, their origin remains poorly understood. Here we show that focal amplifications in breast cancer frequently derive from a mechanism-which we term translocation-bridge amplification-involving inter-chromosomal translocations that lead to dicentric chromosome bridge formation and breakage. In 780 breast cancer genomes, we observe that focal amplifications are frequently connected to each other by inter-chromosomal translocations at their boundaries. Subsequent analysis indicates the following model: the oncogene neighbourhood is translocated in G1 creating a dicentric chromosome, the dicentric chromosome is replicated, and as dicentric sister chromosomes segregate during mitosis, a chromosome bridge is formed and then broken, with fragments often being circularized in extrachromosomal DNAs. This model explains the amplifications of key oncogenes, including ERBB2 and CCND1. Recurrent amplification boundaries and rearrangement hotspots correlate with oestrogen receptor binding in breast cancer cells. Experimentally, oestrogen treatment induces DNA double-strand breaks in the oestrogen receptor target regions that are repaired by translocations, suggesting a role of oestrogen in generating the initial translocations. A pan-cancer analysis reveals tissue-specific biases in mechanisms initiating focal amplifications, with the breakage-fusion-bridge cycle prevalent in some and the translocation-bridge amplification in others, probably owing to the different timing of DNA break repair. Our results identify a common mode of oncogene amplification and propose oestrogen as its mechanistic origin in breast cancer.
© 2023. The Author(s).
Conflict of interest statement
D.P. is a member of the Volastra Therapeutics scientific advisory board.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
