

Senescence-related impairment of autophagy induces toxic intraneuronal amyloid-β accumulation in a mouse model of amyloid pathology
- PMID: 37198698
- PMCID: PMC10189946
- DOI: 10.1186/s40478-023-01578-x
Senescence-related impairment of autophagy induces toxic intraneuronal amyloid-β accumulation in a mouse model of amyloid pathology
Abstract
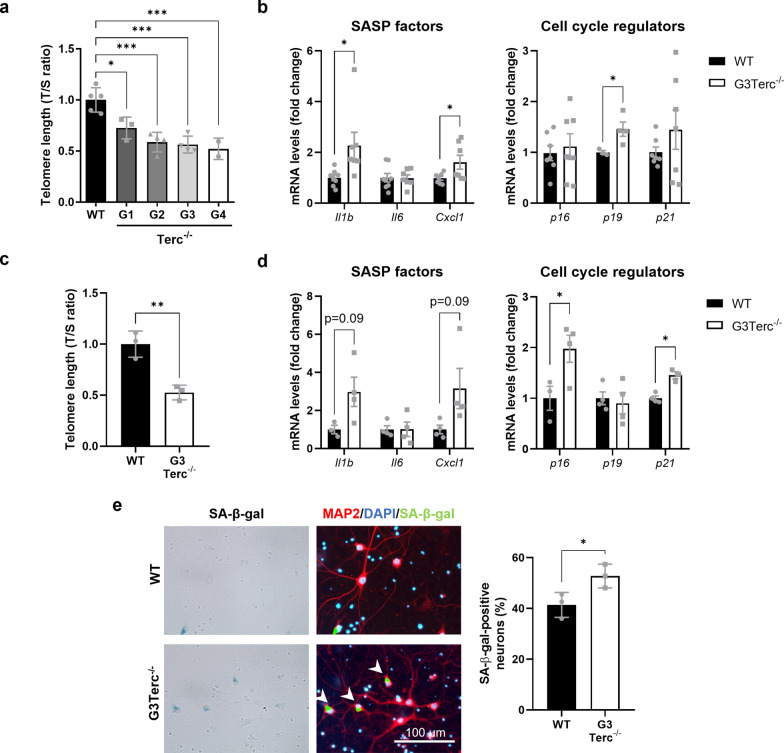
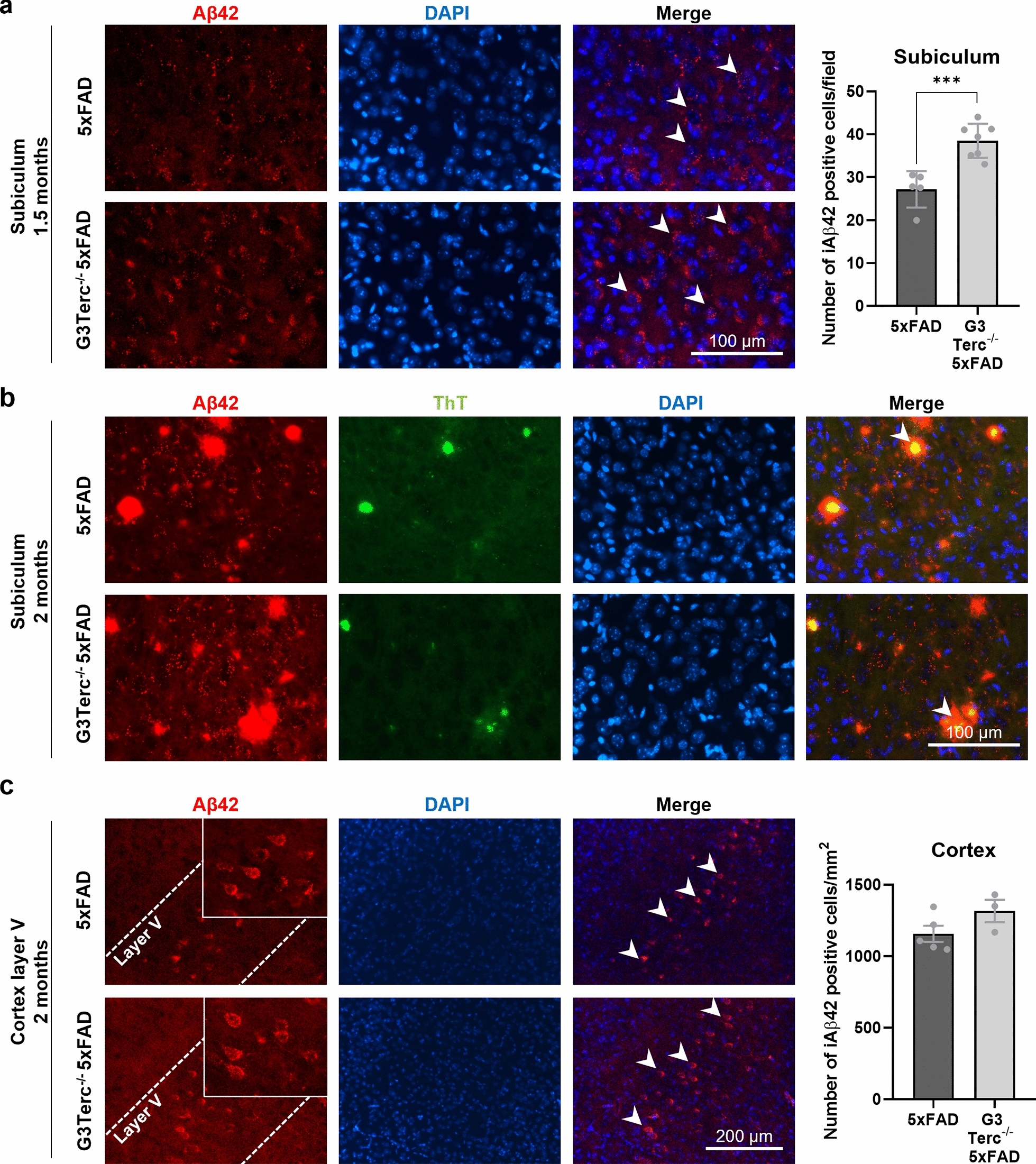
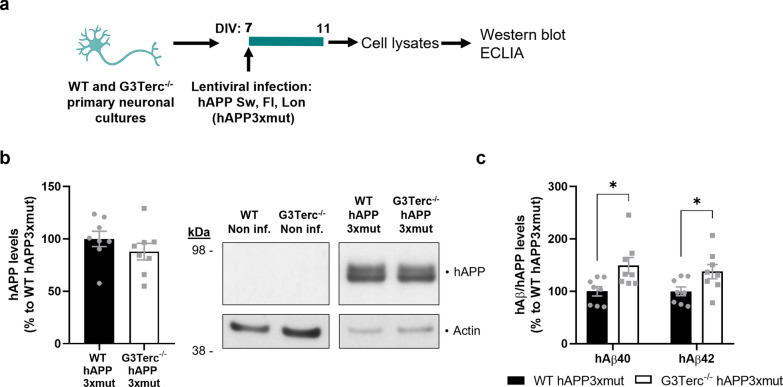
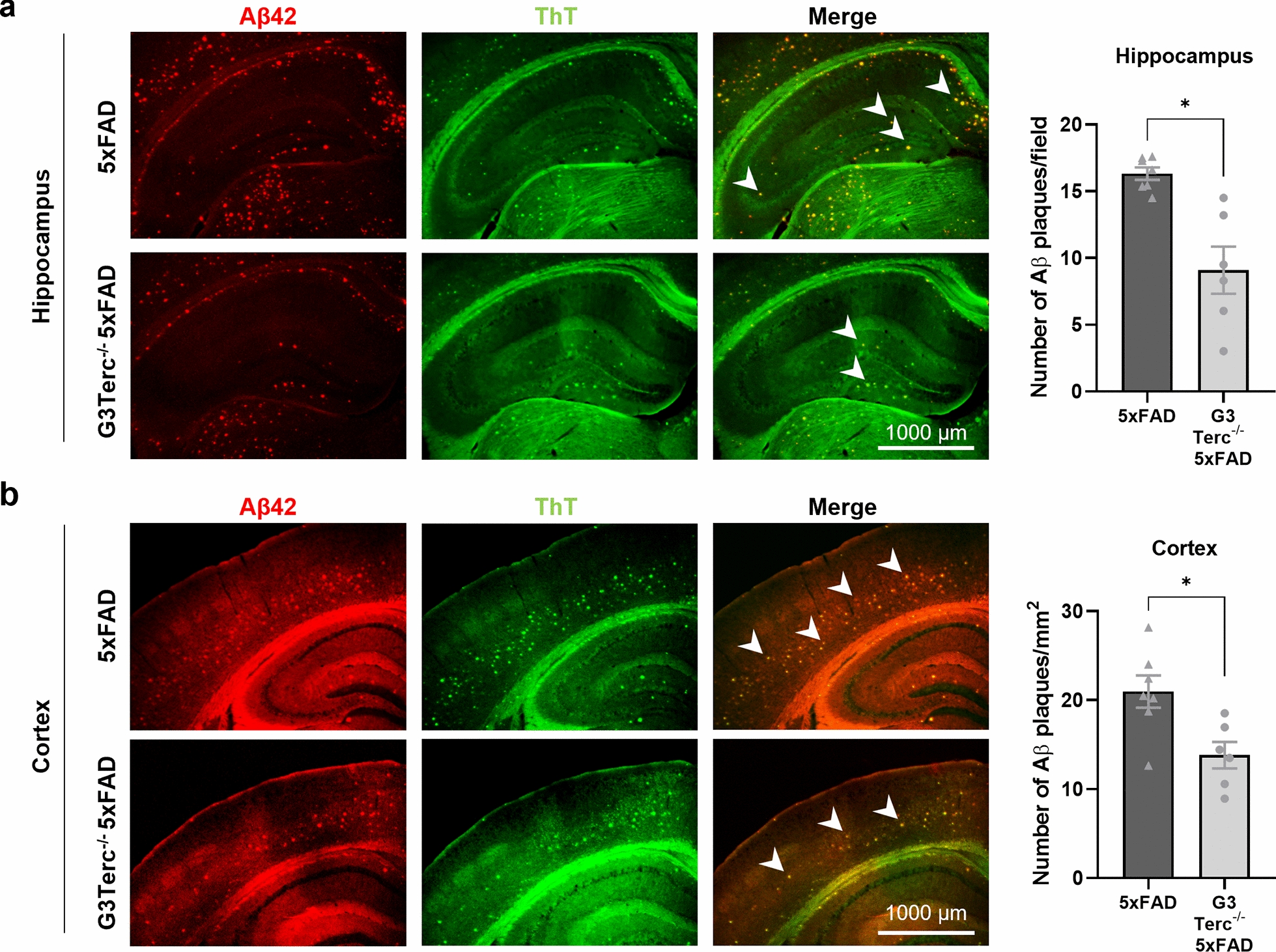
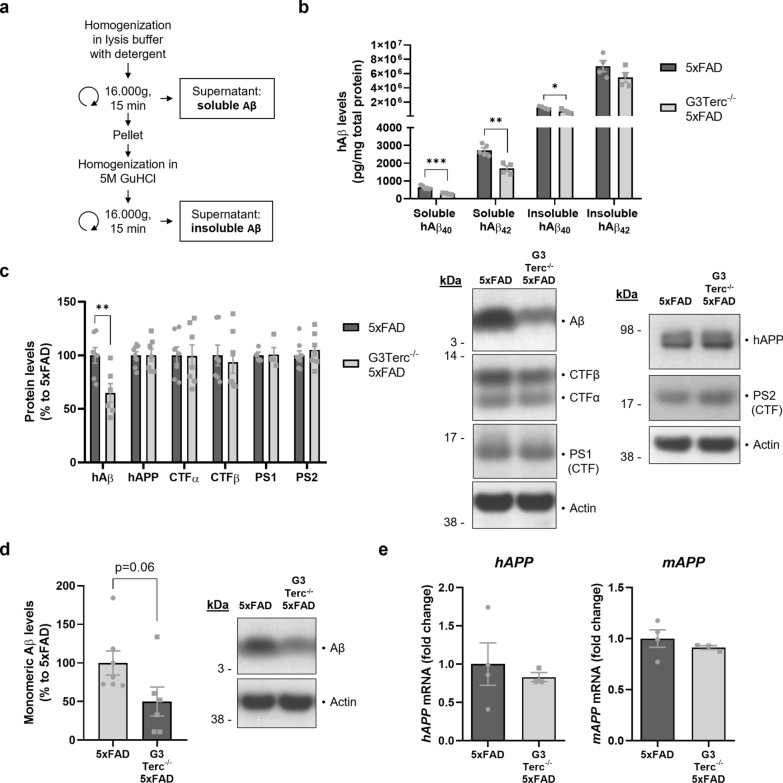
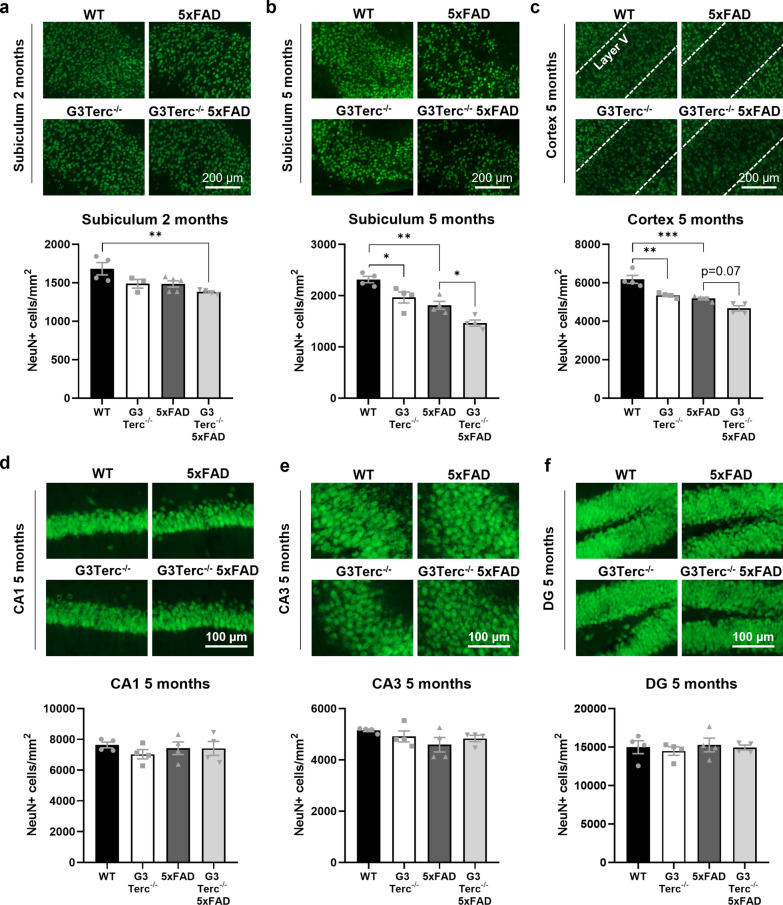
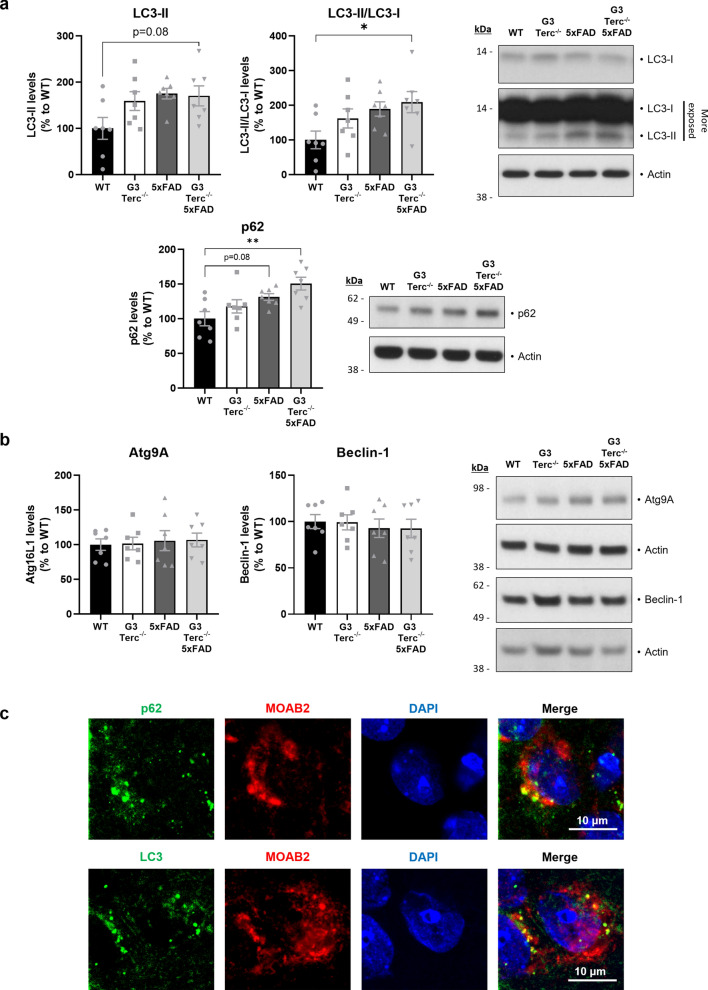
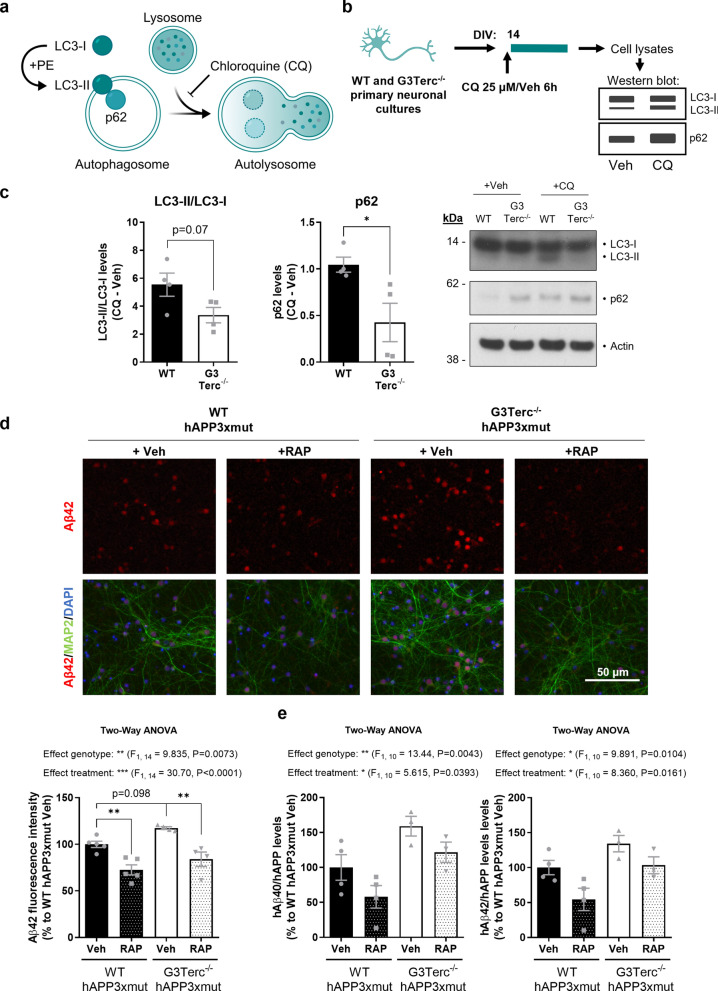
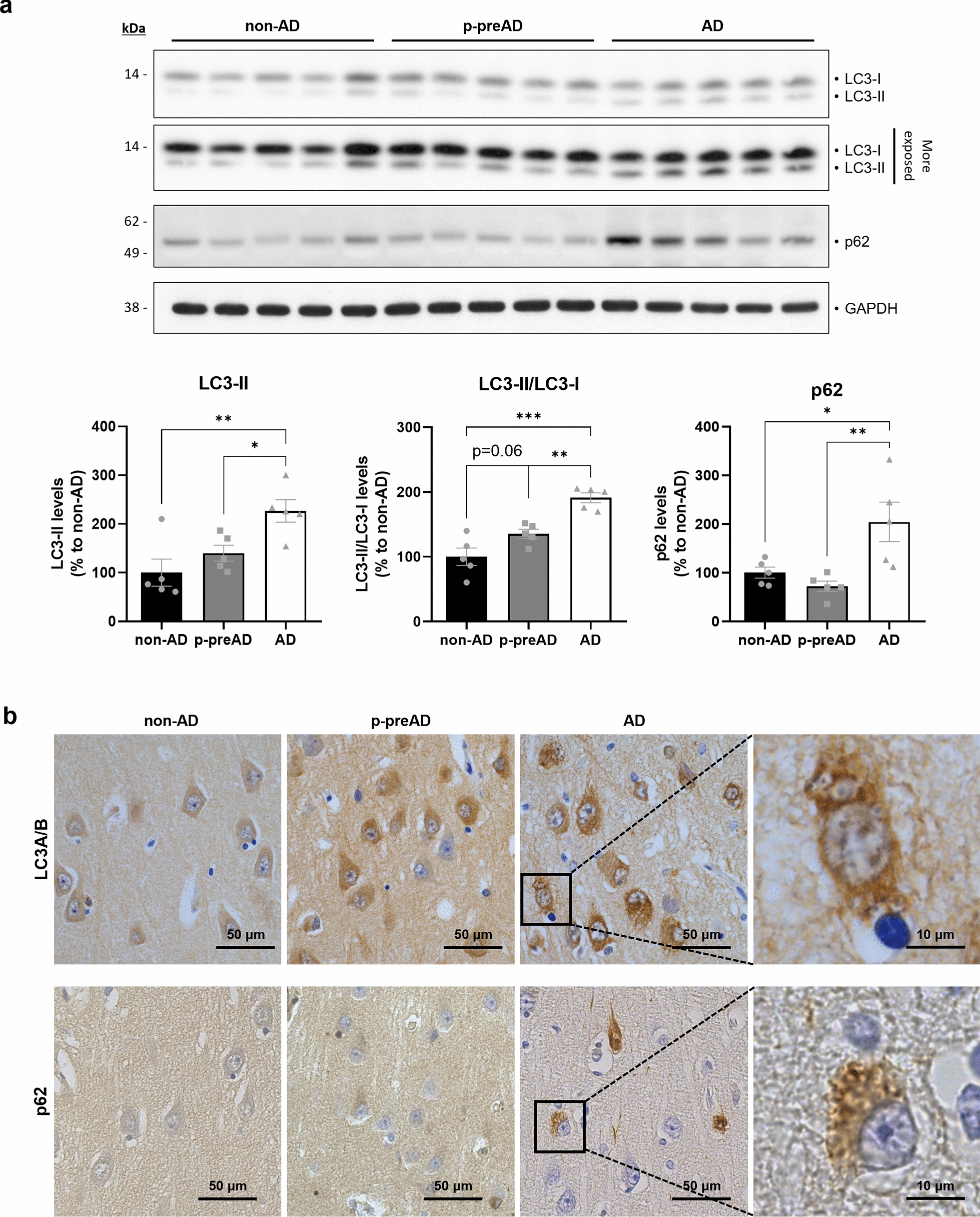
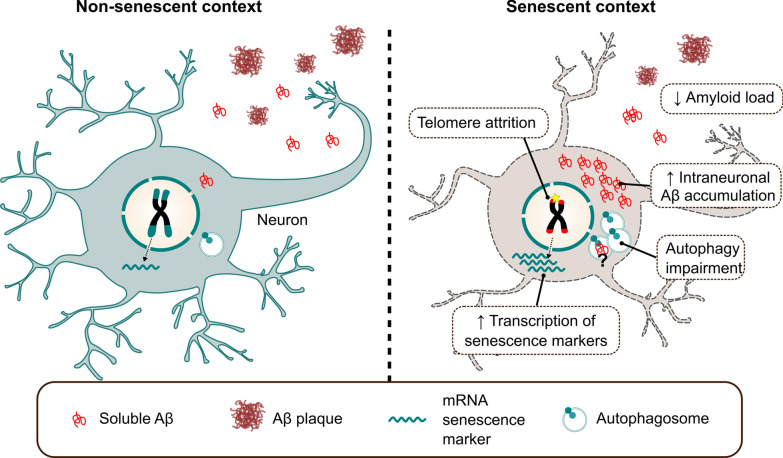
Aging is the main risk factor for Alzheimer's disease (AD) and other neurodegenerative pathologies, but the molecular and cellular changes underlying pathological aging of the nervous system are poorly understood. AD pathology seems to correlate with the appearance of cells that become senescent due to the progressive accumulation of cellular insults causing DNA damage. Senescence has also been shown to reduce the autophagic flux, a mechanism involved in clearing damaged proteins from the cell, and such impairment has been linked to AD pathogenesis. In this study, we investigated the role of cellular senescence on AD pathology by crossing a mouse model of AD-like amyloid-β (Aβ) pathology (5xFAD) with a mouse model of senescence that is genetically deficient for the RNA component of the telomerase (Terc-/-). We studied changes in amyloid pathology, neurodegeneration, and the autophagy process in brain tissue samples and primary cultures derived from these mice by complementary biochemical and immunostaining approaches. Postmortem human brain samples were also processed to evaluate autophagy defects in AD patients. Our results show that accelerated senescence produces an early accumulation of intraneuronal Aβ in the subiculum and cortical layer V of 5xFAD mice. This correlates with a reduction in amyloid plaques and Aβ levels in connecting brain regions at a later disease stage. Neuronal loss was specifically observed in brain regions presenting intraneuronal Aβ and was linked to telomere attrition. Our results indicate that senescence affects intraneuronal Aβ accumulation by impairing autophagy function and that early autophagy defects can be found in the brains of AD patients. Together, these findings demonstrate the instrumental role of senescence in intraneuronal Aβ accumulation, which represents a key event in AD pathophysiology, and emphasize the correlation between the initial stages of amyloid pathology and defects in the autophagy flux.
Keywords: Alzheimer’s disease; Autophagy; Cellular senescence; Intraneuronal Aβ; Telomere shortening.
© 2023. The Author(s).
Conflict of interest statement
DRT received speaker honorary or travel reimbursement from UCB (Brussels, Belgium) and Biogen (USA), and collaborated with Novartis Pharma AG (Basel, Switzerland), Probiodrug (Halle (Saale), Germany), GE Healthcare (Amersham, UK), and Janssen Pharmaceutical Companies (Beerse, Belgium). RV’s institution has clinical trial agreements (RV as Principal Investigator) with Alector, Biogen, Denali, J&J, Prevail, UCB, Roche and Wave. RV’s institution has consultancy agreements for DSMB membership (RV) with AC Immune and Novartis. CAFvA received honoraria from serving on the scientific advisory board of Biogen, Roche, Novo Nordisk, and Dr. Wilmar Schwabe GmbH &Co. KG, funding for travel and speaker honoraria from Biogen, Roche diagnostics AG, Novartis, Medical Tribune Verlagsgesellschaft GmbH, Landesvereinigung für Gesundheit und Akademie für Sozialmedizin Niedersachsen e. V. and Dr. Wilmar Schwabe GmbH &Co. KG and has received research support from Roche diagnostics AG. The other authors have no relevant financial or non-financial interests to disclose.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
