

Relationship of paroxysmal nocturnal hemoglobinuria (PNH) granulocyte clone size to disease burden and risk of major vascular events in untreated patients: results from the International PNH Registry
- PMID: 37199789
- PMCID: PMC10261189
- DOI: 10.1007/s00277-023-05269-4
Relationship of paroxysmal nocturnal hemoglobinuria (PNH) granulocyte clone size to disease burden and risk of major vascular events in untreated patients: results from the International PNH Registry
Abstract
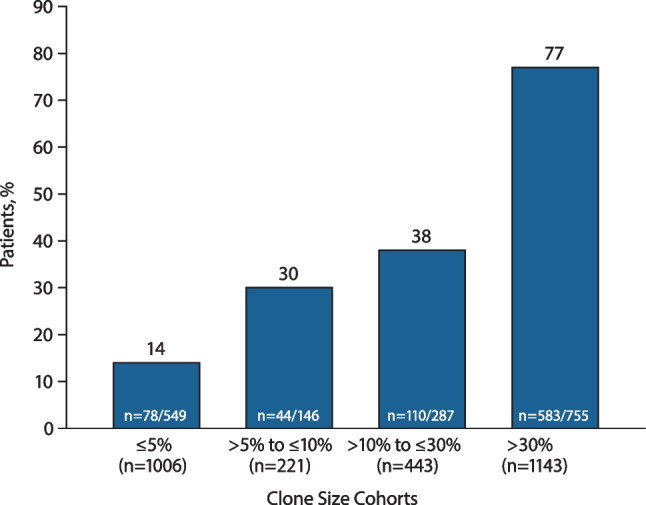
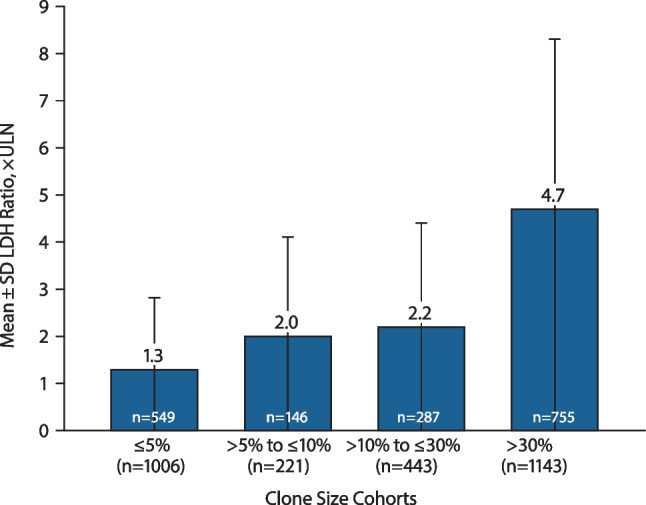
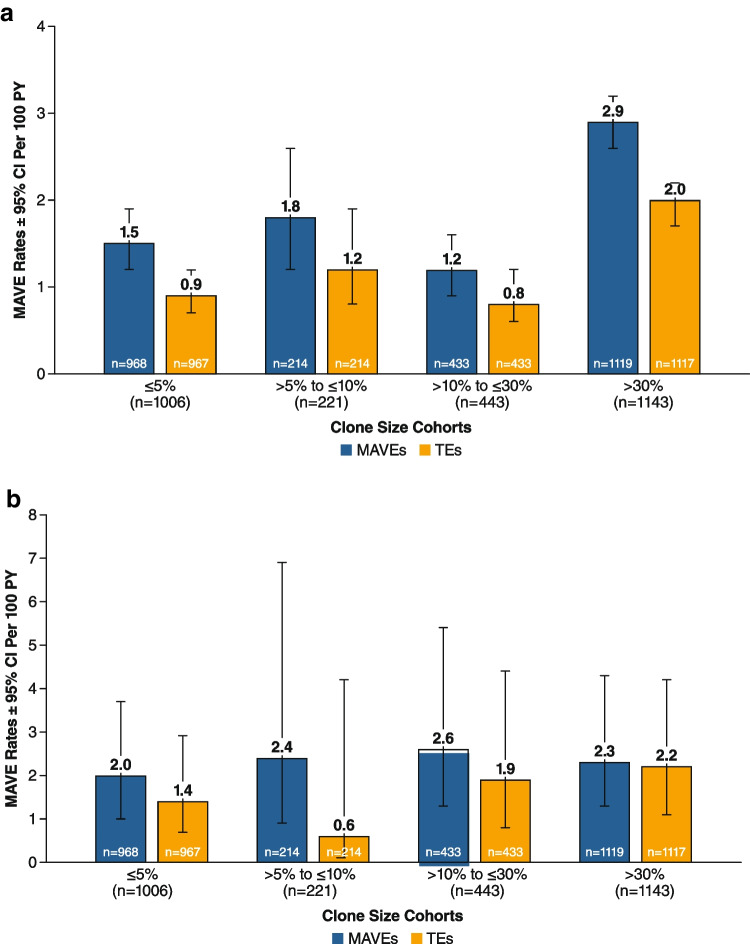
Paroxysmal nocturnal hemoglobinuria (PNH) is caused by acquired gene mutations resulting in deficiency of glycosylphosphatidylinositol (GPI)-anchored complement regulatory proteins on the surface of blood cells, leading to terminal complement-mediated intravascular hemolysis and increased risk of major adverse vascular events (MAVEs). Using data from the International PNH Registry, this study investigated the relationship between the proportion of GPI-deficient granulocytes at PNH onset and (1) the risk for MAVEs (including thrombotic events [TEs]) and (2) the following parameters at last follow-up: high disease activity (HDA); lactate dehydrogenase (LDH) ratio; fatigue; abdominal pain; and rates of overall MAVEs and TEs. A total of 2813 patients untreated at enrollment were included and stratified by clone size at PNH disease onset (baseline). At last follow-up, higher proportion of GPI-deficient granulocytes (≤ 5% vs. > 30% clone size) at baseline was associated with significantly increased HDA incidence (14% vs. 77%), mean LDH ratio (1.3 vs. 4.7 × upper limit of normal), and rates of MAVEs 1.5 vs. 2.9 per 100 person-years) and TEs (0.9 vs. 2.0 per 100 person-years). Fatigue was evident in 71 to 76% of patients regardless of clone size. Abdominal pain was more frequently reported with clone size > 30%. A larger clone size at baseline appears to indicate a greater disease burden and risk of TEs and MAVEs and may inform decision making among physicians managing PNH patients at risk of experiencing TEs or other MAVEs. ClinicalTrials.gov ID: NCT01374360.
Keywords: Cohort studies; Disease progression; GPI-deficient granulocytes, Risk factors; Paroxysmal nocturnal hemoglobinuria; Thromboembolism.
© 2023. The Author(s).
Conflict of interest statement
David Dingli has received honoraria and consulting fees from Alexion, AstraZeneca Rare Disease, Apellis, Novartis and Sanofi, and participates in the International PNH Registry (for Mayo Clinic, Rochester) for Alexion, AstraZeneca Rare Disease. Jaroslaw P. Maciejewski has received consulting fees from Alexion, AstraZeneca Rare Disease; Apellis Pharmaceuticals; and Ra Pharma. He has also received speaker fees from and is a member of the Executive Committee of the International PNH Registry for Alexion, AstraZeneca Rare Disease. Loree Larratt has received honoraria and research support (for the laboratory at the University of Alberta, for ADAMTS13 testing) from Alexion, AstraZeneca Rare Disease. Ronald S. Go participates in the International PNH Registry (for Mayo Clinic, Rochester) for Alexion, AstraZeneca Rare Disease. Britta Höchsmann has received honoraria, consulting fees, and research support (to University Hospital of Ulm) from Alexion, AstraZeneca Rare Disease, Novartis, Apellis Pharmaceuticals, Sobi, and Roche. Ke Zu was an employee of Alexion, AstraZeneca Rare Disease at the time of the study, and is currently affiliated with Merck & Co., Inc. Philippe Gustovic is an employee and stockholder of Alexion, AstraZeneca Rare Disease. Alexander D. Kulagin has received honoraria and consulting fees from Alexion, AstraZeneca Rare Disease, Novartis, and GENERIUM JSC, and is a member of the Executive Committee of the International PNH Registry for Alexion, AstraZeneca Rare Disease.
Figures
References
-
- Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socie G, International P. N. H. Interest Group Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–3709. doi: 10.1182/blood-2005-04-1717. - DOI - PMC - PubMed
-
- Richards SJ, Painter D, Dickinson AJ, Griffin M, Munir T, Arnold L, Payne D, Pike A, Muus P, Hill A, Newton DJ, McKinley C, Jones R, Kelly R, Smith A, Roman E, Hillmen P. The incidence and prevalence of patients with paroxysmal nocturnal haemoglobinuria and aplastic anaemia PNH syndrome: a retrospective analysis of the UK’s population-based haematological malignancy research network 2004–2018. Eur J Haematol. 2021;107(2):211–218. doi: 10.1111/ejh.13640. - DOI - PubMed
-
- Jalbert JJ, Chaudhari U, Zhang H, Weyne J, Shammo JM (2019) Epidemiology of PNH and real-world treatment patterns following an incident PNH diagnosis in the US. Blood 134(Supplement_1):3407–3407. 10.1182/blood-2019-125867
-
- Socie G, Schrezenmeier H, Muus P, Lisukov I, Roth A, Kulasekararaj A, Lee JW, Araten D, Hill A, Brodsky R, Urbano-Ispizua A, Szer J, Wilson A, Hillmen P, Registry PNH. Changing prognosis in paroxysmal nocturnal haemoglobinuria disease subcategories: an analysis of the International PNH Registry. Intern Med J. 2016;46(9):1044–1053. doi: 10.1111/imj.13160. - DOI - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
