

Within-patient and global evolutionary dynamics of Klebsiella pneumoniae ST17
- PMID: 37200066
- PMCID: PMC10272876
- DOI: 10.1099/mgen.0.001005
Within-patient and global evolutionary dynamics of Klebsiella pneumoniae ST17
Abstract
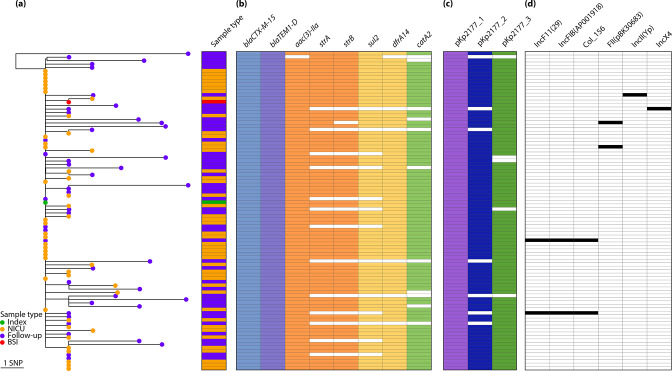
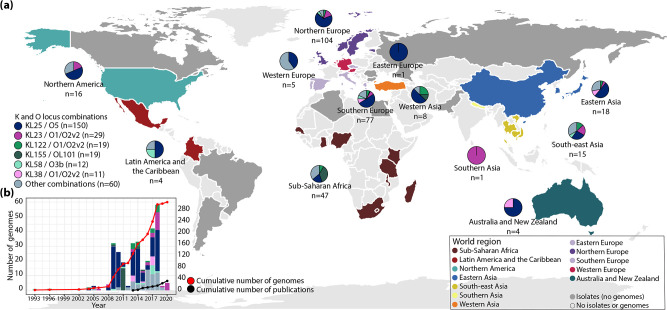
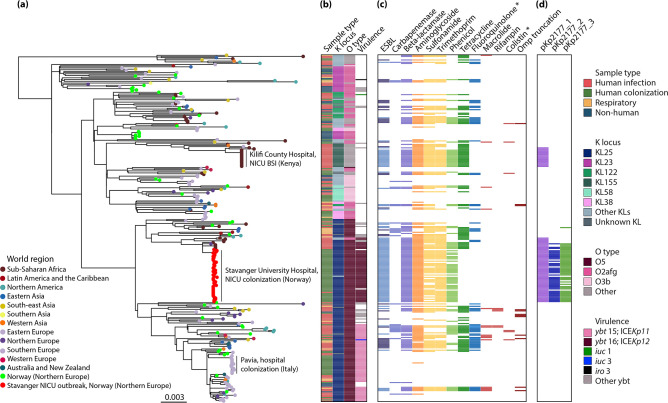
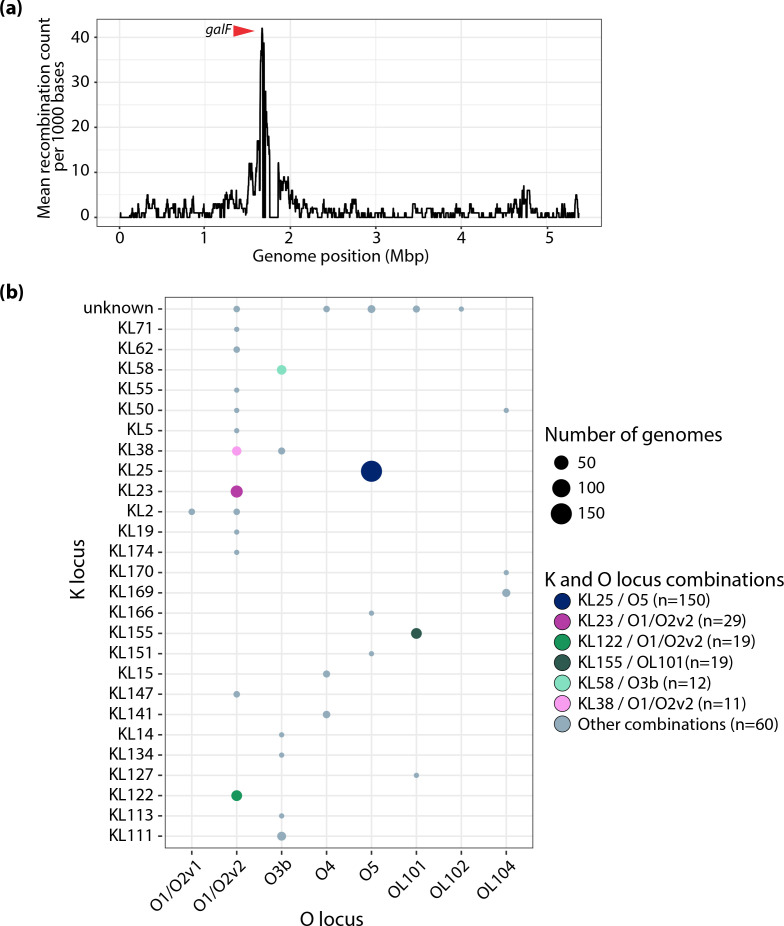
Klebsiella pneumoniae sequence type (ST) 17 is a global problem clone that causes multidrug-resistant (MDR) hospital infections worldwide. In 2008-2009, an outbreak of MDR ST17 occurred at a neonatal intensive care unit (NICU) in Stavanger, Norway. Fifty-seven children were colonized. We observed intestinal persistence of ST17 in all of the children for up to two years after hospital discharge. Here, we investigated the within-host evolution of ST17 in 45 of those children during long-term colonization and compared the outbreak with 254 global strains. Ninety-two outbreak-related isolates were whole-genome sequenced. They had capsule locus KL25, O locus O5 and carried yersiniabactin. During within-host colonization ST17 remained stable with few single nucleotide polymorphisms, no acquisition of antimicrobial resistance (AMR) or virulence determinants, and persistent carriage of a bla CTX-M-15-encoding IncFII(K) IncFIB(K) plasmid (pKp2177_1). The global collection included ST17 from 1993 to 2020 from 34 countries, that were from human infection (41.3%), colonization (39.3%) and respiratory specimens (7.3%), from animals (9.3%), and from the environment (2.7%). We estimate that ST17 emerged mid-to-late 19th century (1859, 95 % HPD 1763-1939) and diversified through recombinations of the K and O loci to form several sublineages, with various AMR genes, virulence loci and plasmids. There was limited evidence of persistence of AMR genes in any of these lineages. A globally disseminated sublineage with KL25/O5 accounted for 52.7 % of the genomes. It included a monophyletic subclade that emerged in the mid-1980s, which comprised the Stavanger NICU outbreak and 10 genomes from three other countries, which all carried pKp2177_1. The plasmid was also observed in a KL155/OL101 subclade from the 2000s. Three clonal expansions of ST17 were identified; all were healthcare-associated and carried either yersiniabactin and/or pKp2177_1. To conclude, ST17 is globally disseminated and associated with opportunistic hospital-acquired infections. It contributes to the burden of global MDR infections, but many diverse lineages persist without acquired AMR. We hypothesize that non-human sources and human colonization may play a crucial role for severe infections in vulnerable patients, such as preterm neonates.
Keywords: Klebsiella pneumoniae; ST17; colonization; global dynamics; in vivo evolution; infection.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
-
- Sonda T, Kumburu H, van Zwetselaar M, Alifrangis M, Mmbaga BT, et al. Molecular epidemiology of virulence and antimicrobial resistance determinants in Klebsiella pneumoniae from hospitalised patients in Kilimanjaro, Tanzania. Eur J Clin Microbiol Infect Dis. 2018;37:1901–1914. doi: 10.1007/s10096-018-3324-5. - DOI - PubMed
-
- Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci. 2015;112:E3574–81. doi: 10.1073/pnas.1501049112. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources