

The Analytical Flory Random Coil Is a Simple-to-Use Reference Model for Unfolded and Disordered Proteins
- PMID: 37200094
- PMCID: PMC10875986
- DOI: 10.1021/acs.jpcb.3c01619
The Analytical Flory Random Coil Is a Simple-to-Use Reference Model for Unfolded and Disordered Proteins
Abstract
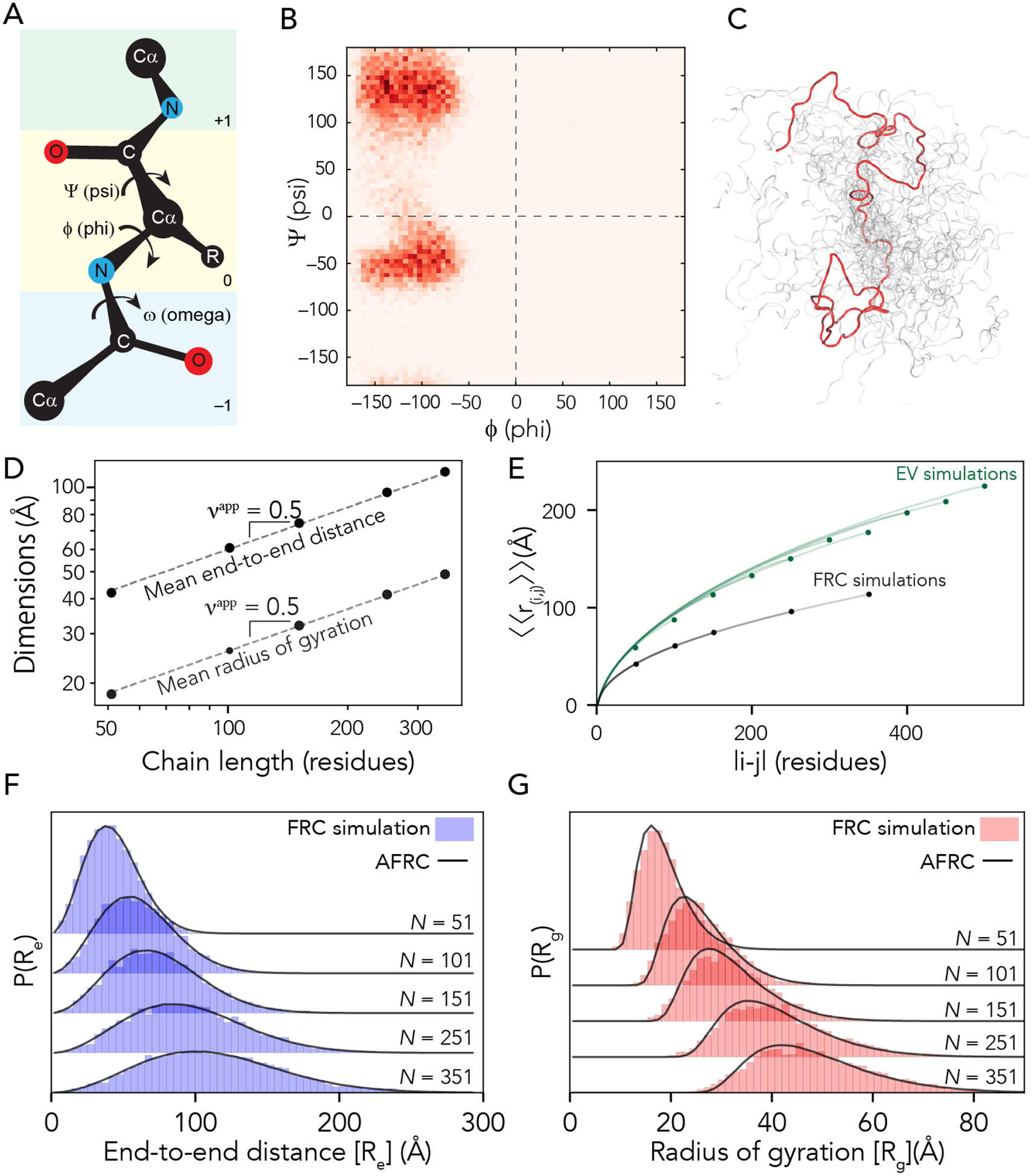
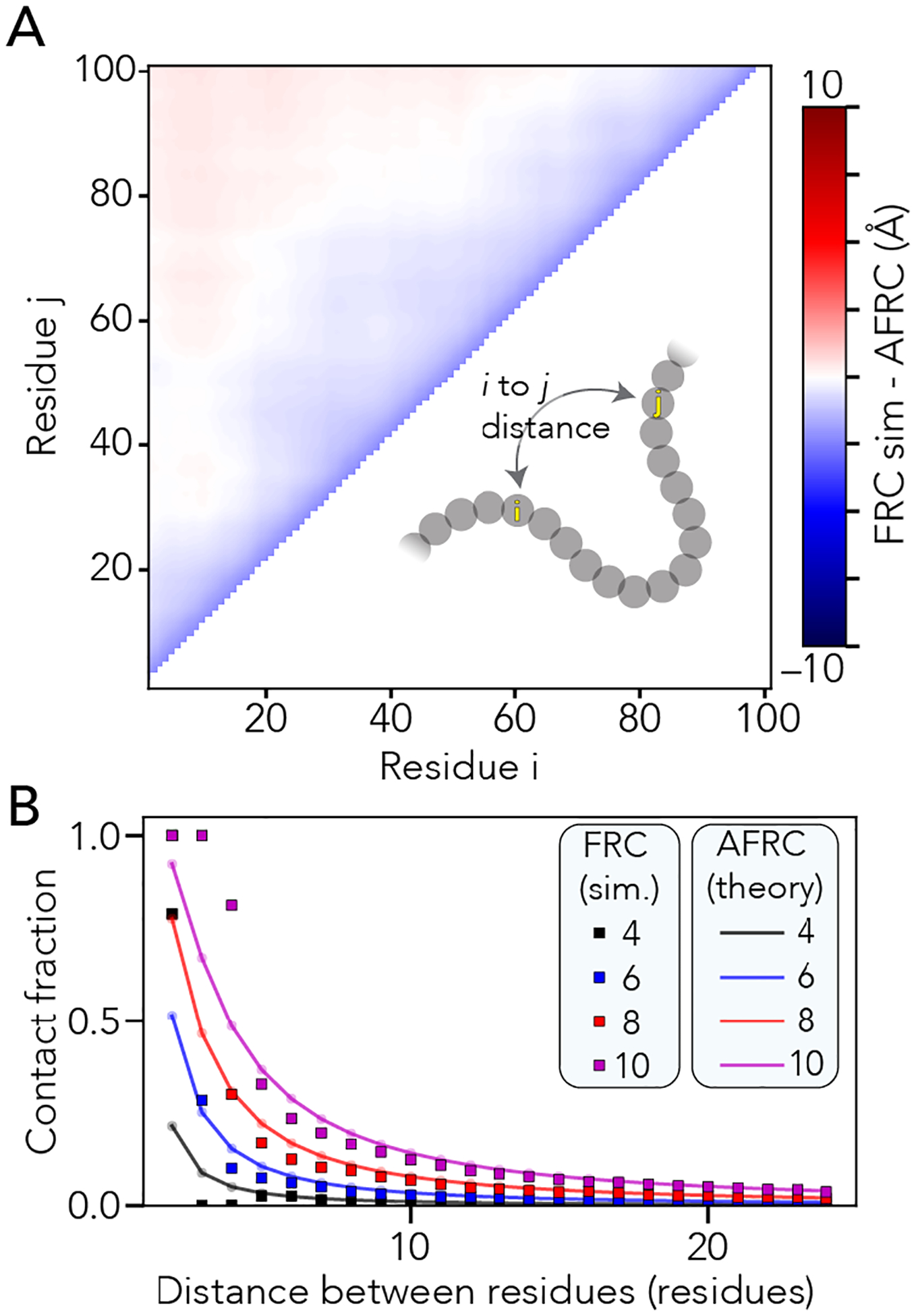
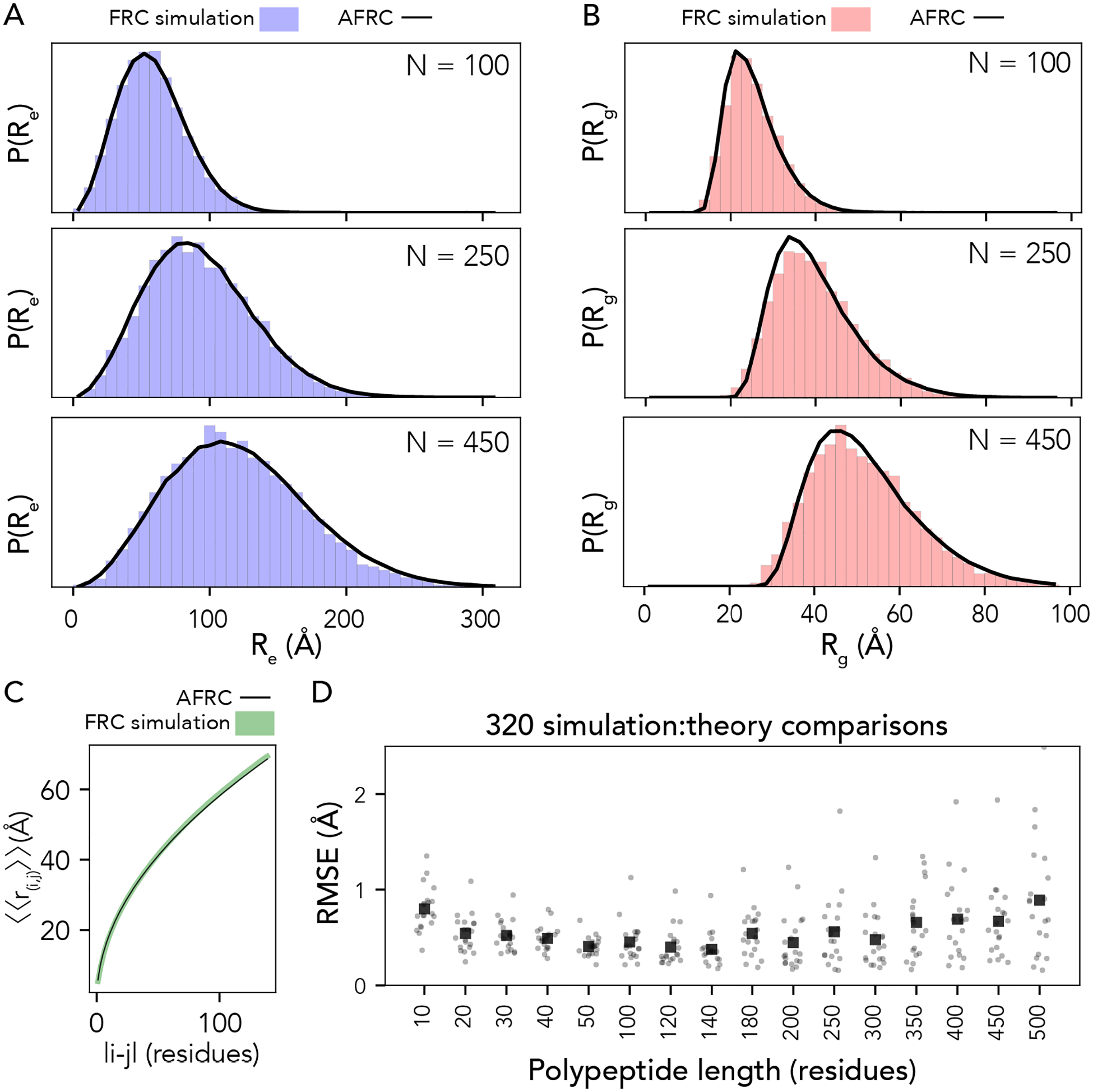
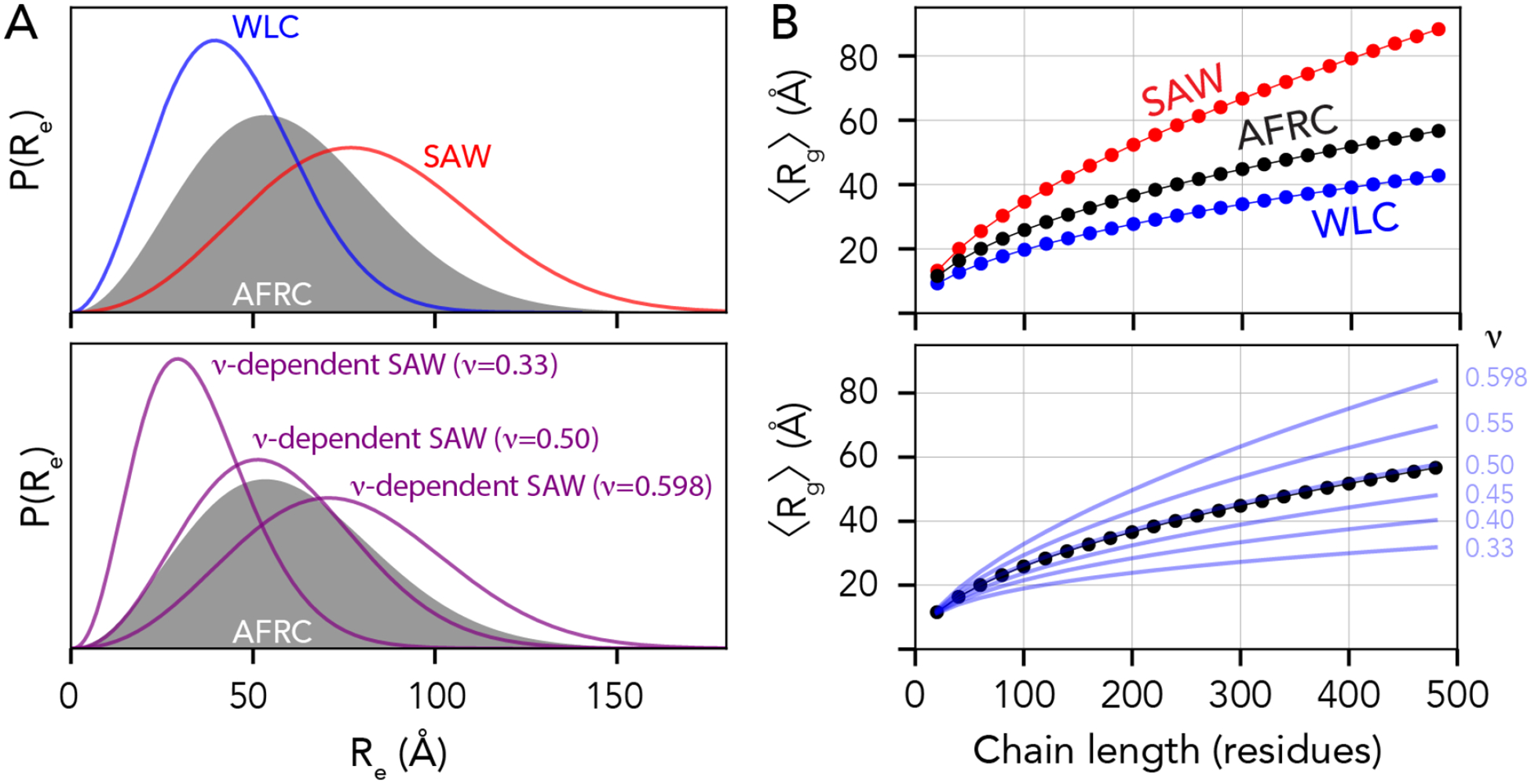
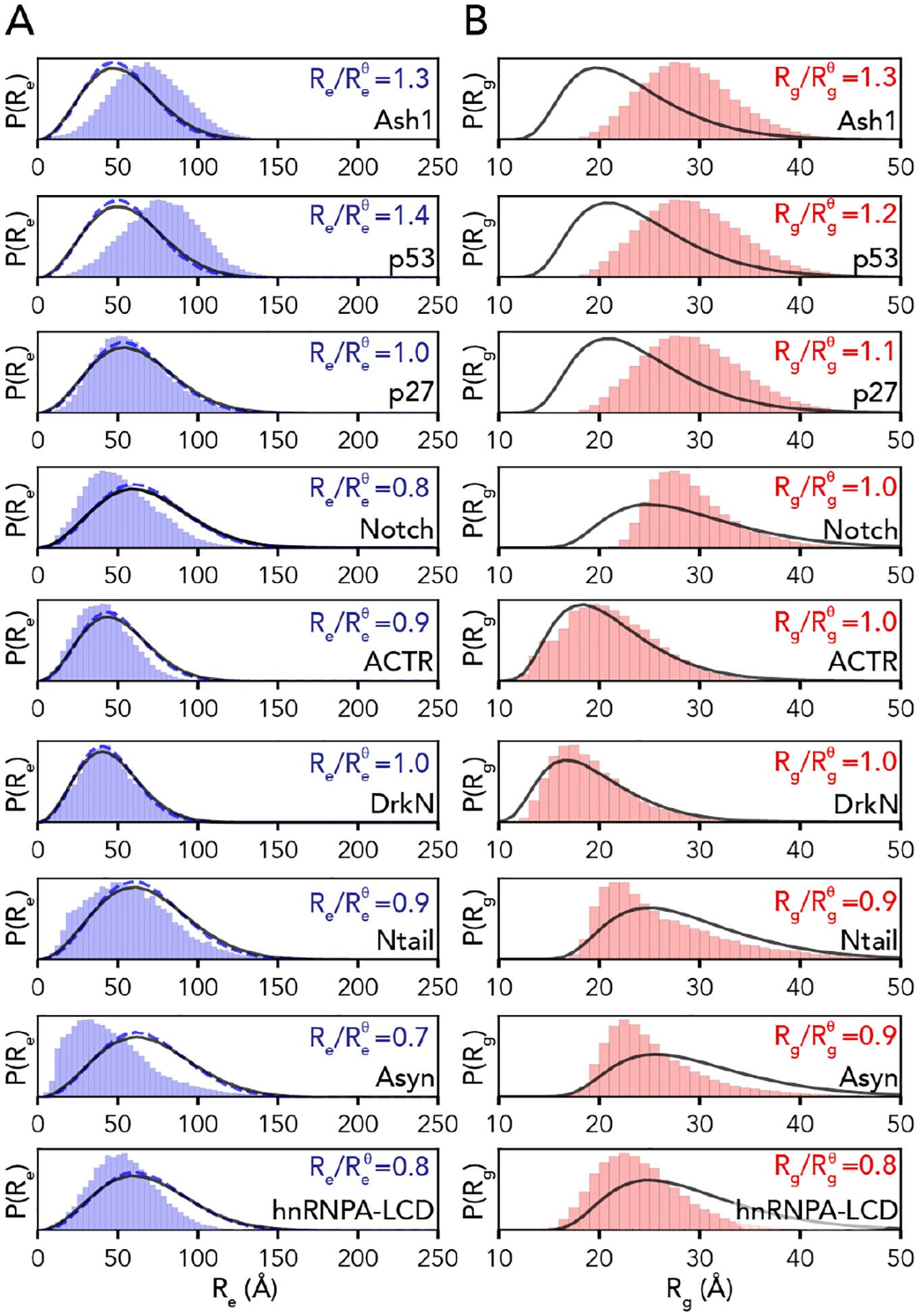
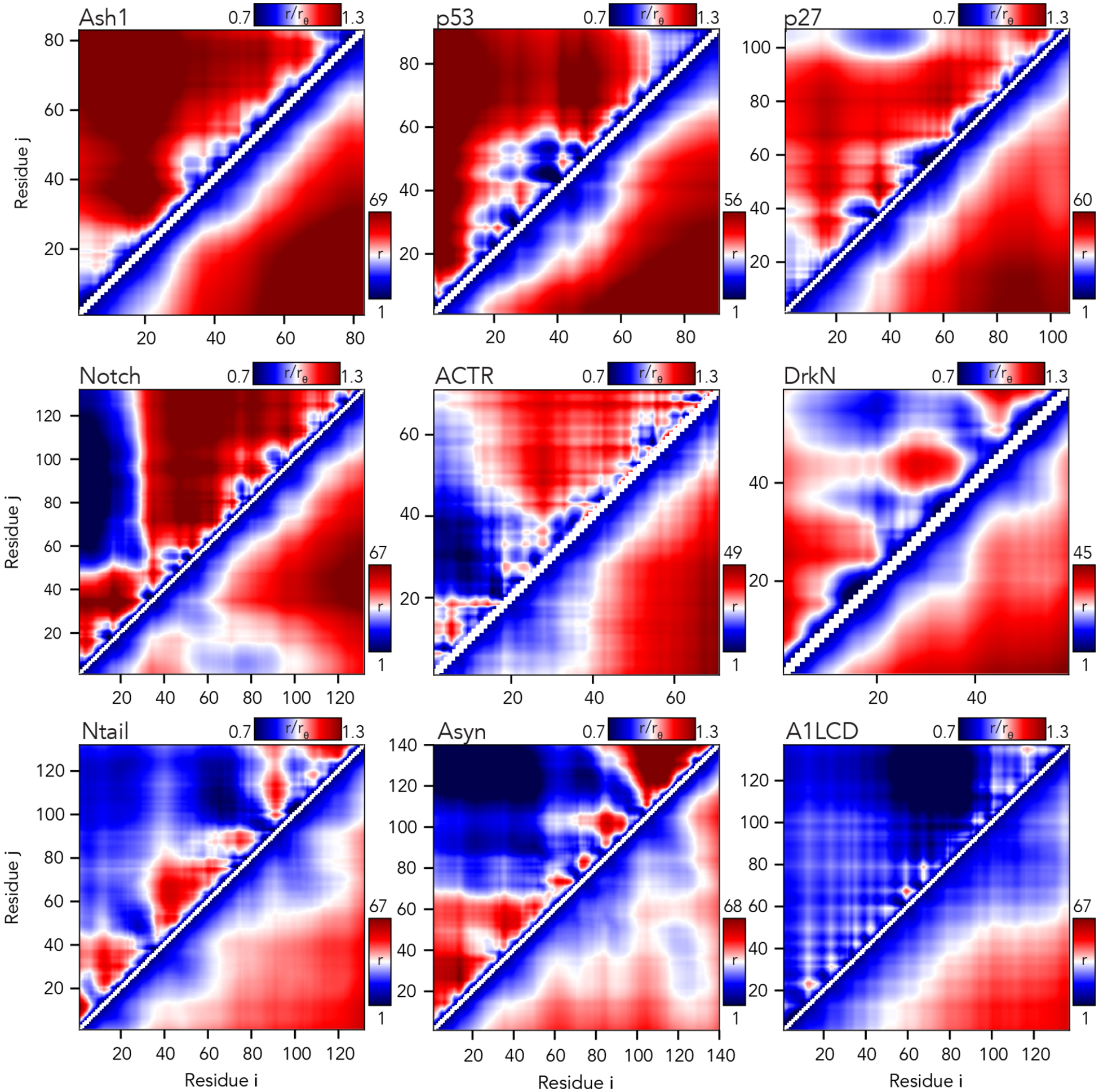
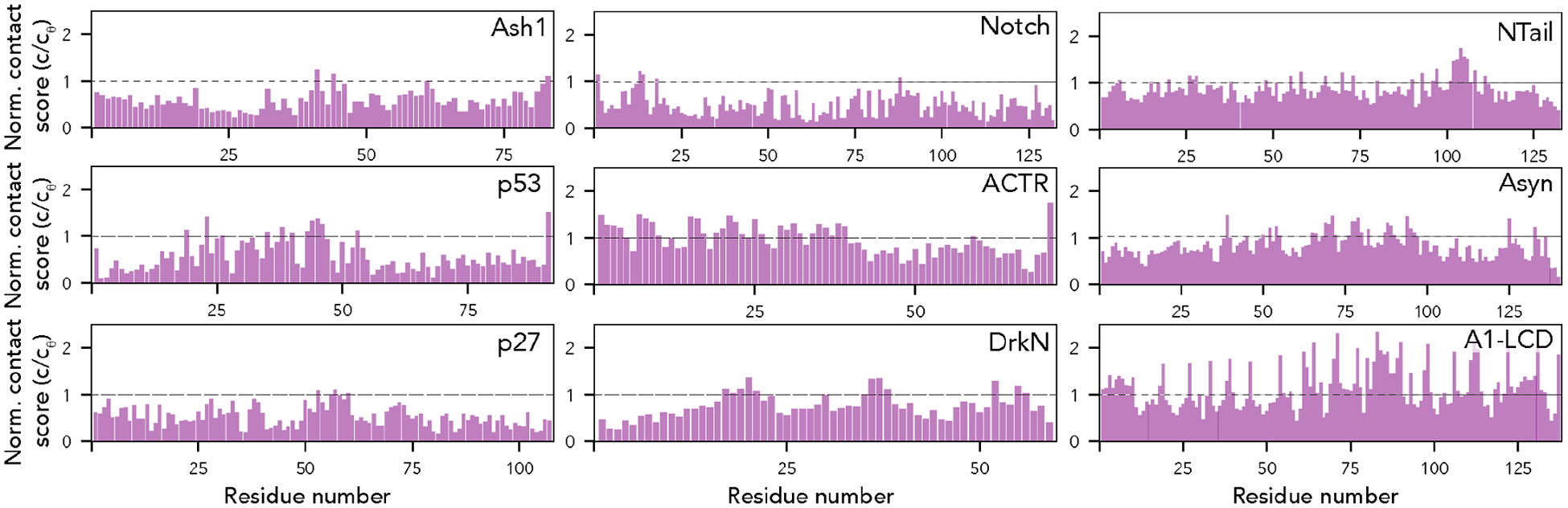
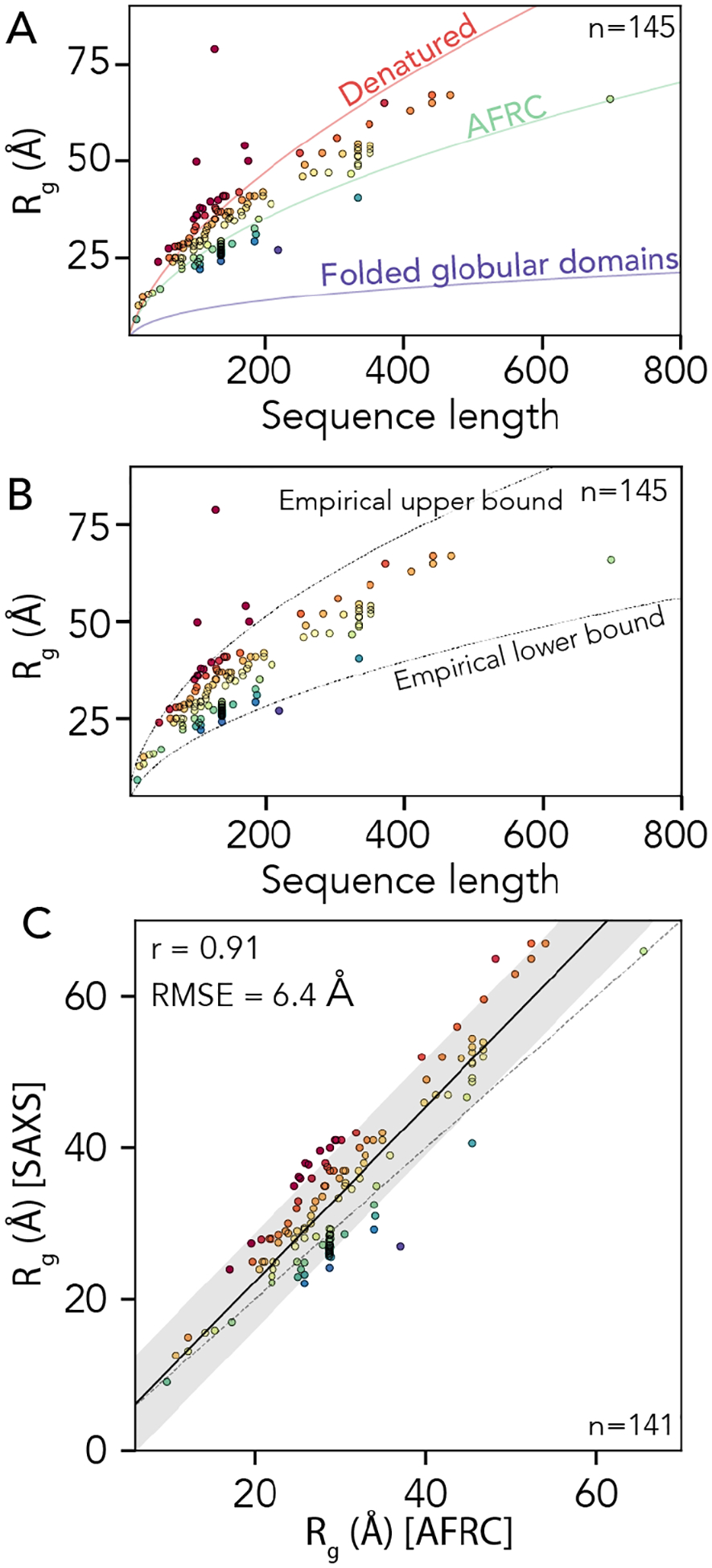
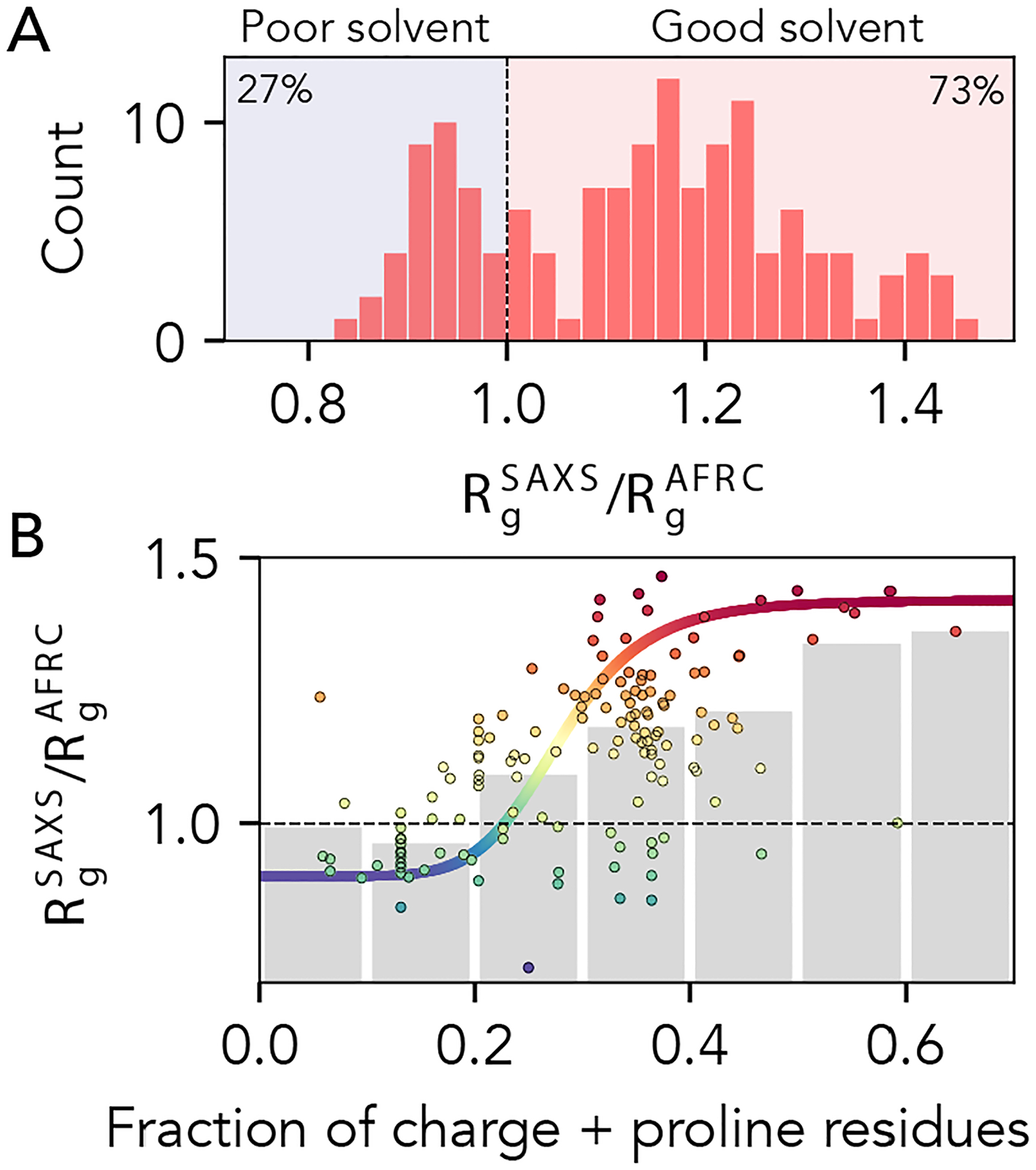
Denatured, unfolded, and intrinsically disordered proteins (collectively referred to here as unfolded proteins) can be described using analytical polymer models. These models capture various polymeric properties and can be fit to simulation results or experimental data. However, the model parameters commonly require users' decisions, making them useful for data interpretation but less clearly applicable as stand-alone reference models. Here we use all-atom simulations of polypeptides in conjunction with polymer scaling theory to parameterize an analytical model of unfolded polypeptides that behave as ideal chains (ν = 0.50). The model, which we call the analytical Flory random coil (AFRC), requires only the amino acid sequence as input and provides direct access to probability distributions of global and local conformational order parameters. The model defines a specific reference state to which experimental and computational results can be compared and normalized. As a proof-of-concept, we use the AFRC to identify sequence-specific intramolecular interactions in simulations of disordered proteins. We also use the AFRC to contextualize a curated set of 145 different radii of gyration obtained from previously published small-angle X-ray scattering experiments of disordered proteins. The AFRC is implemented as a stand-alone software package and is also available via a Google Colab notebook. In summary, the AFRC provides a simple-to-use reference polymer model that can guide intuition and aid in interpreting experimental or simulation results.
Figures
Update of
-
The analytical Flory random coil is a simple-to-use reference model for unfolded and disordered proteins.bioRxiv [Preprint]. 2023 Mar 13:2023.03.12.531990. doi: 10.1101/2023.03.12.531990. bioRxiv. 2023. Update in: J Phys Chem B. 2023 Jun 1;127(21):4746-4760. doi: 10.1021/acs.jpcb.3c01619. PMID: 36993592 Free PMC article. Updated. Preprint.
References
-
- Dill KA; Shortle D Denatured States of Proteins. Annu. Rev. Biochem 1991, 60, 795–825. - PubMed
-
- Chan HS; Dill KA Polymer Principles in Protein Structure and Stability. Annu. Rev. Biophys. Biophys. Chem 1991, 20, 447–490. - PubMed
-
- Schuler B; Soranno A; Hofmann H; Nettels D Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys 2016, 45, 207–231. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
